Mavridou Dimitra, Psatha Konstantina, Aivaliotis Michalis
Laboratory of Biological Chemistry, School of Medicine, Faculty of Health Sciences, Aristotle University of Thessaloniki, GR-54124 Thessaloniki, Greece.
Functional Proteomics and Systems Biology (FunPATh), Center for Interdisciplinary Research and Innovation (CIRI-AUTH), GR-57001 Thessaloniki, Greece.
J Pers Med. 2024 Aug 6;14(8):831. doi: 10.3390/jpm14080831.
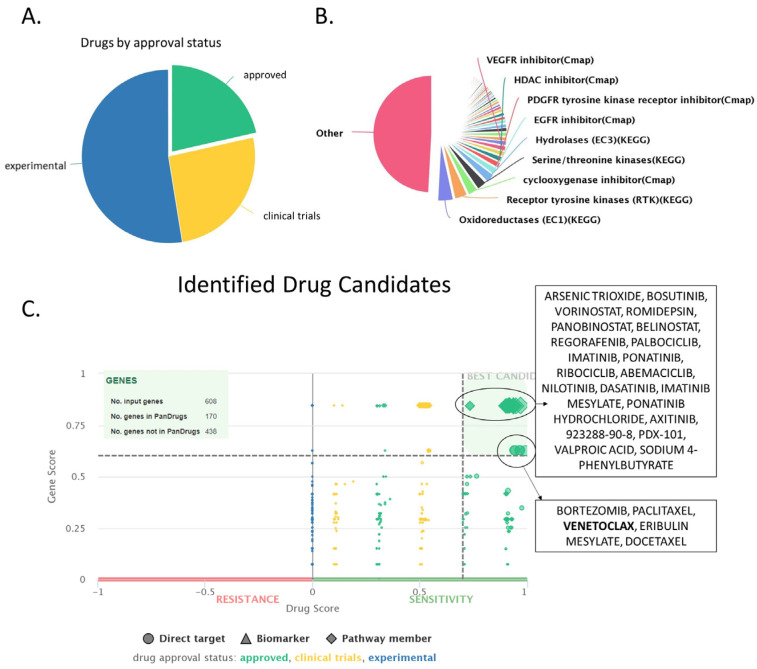
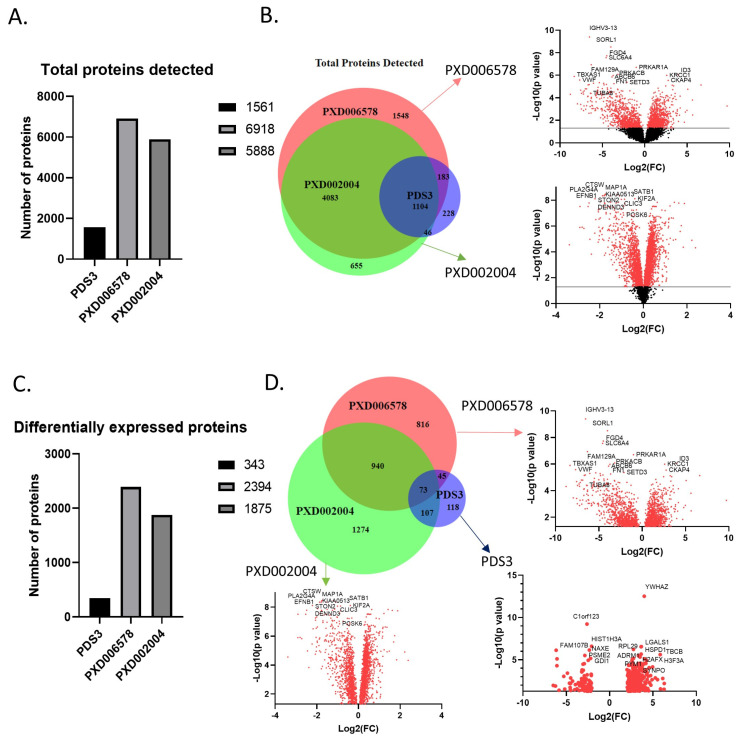
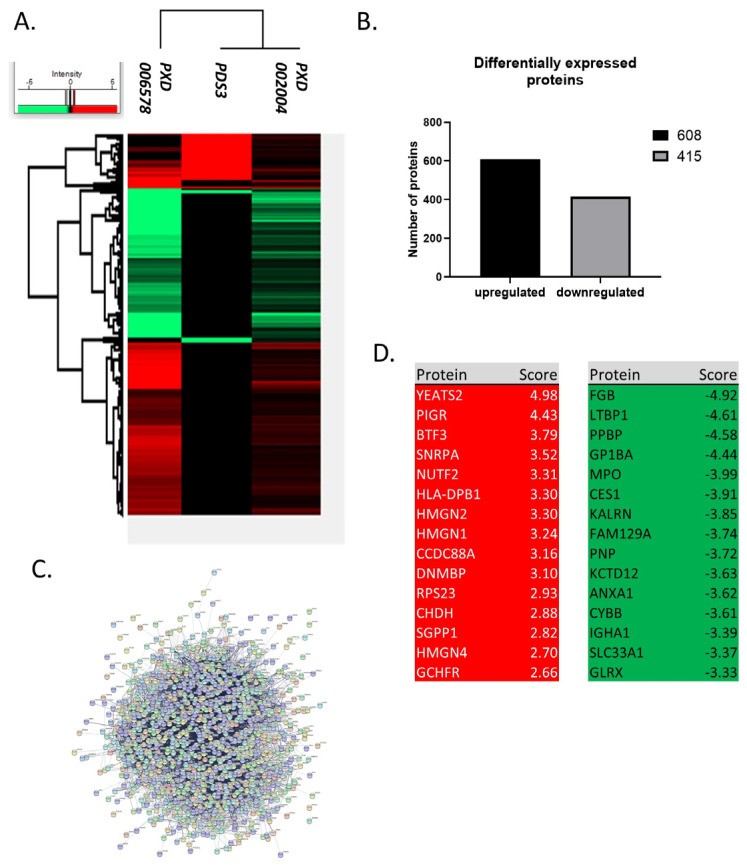
Chronic Lymphocytic Leukemia (CLL) is the most common B-cell malignancy in the Western world, characterized by frequent relapses despite temporary remissions. Our study integrated publicly available proteomic, transcriptomic, and patient survival datasets to identify key differences between healthy and CLL samples. We exposed approximately 1000 proteins that differentiate healthy from cancerous cells, with 608 upregulated and 415 downregulated in CLL cases. Notable upregulated proteins include YEATS2 (an epigenetic regulator), PIGR (Polymeric immunoglobulin receptor), and SNRPA (a splicing factor), which may serve as prognostic biomarkers for this disease. Key pathways implicated in CLL progression involve RNA processing, stress resistance, and immune response deficits. Furthermore, we identified three existing drugs-Bosutinib, Vorinostat, and Panobinostat-for potential further investigation in drug repurposing in CLL. We also found limited correlation between transcriptomic and proteomic data, emphasizing the importance of proteomics in understanding gene expression regulation mechanisms. This generally known disparity highlights once again that mRNA levels do not accurately predict protein abundance due to many regulatory factors, such as protein degradation, post-transcriptional modifications, and differing rates of translation. These results demonstrate the value of integrating omics data to uncover deregulated proteins and pathways in cancer and suggest new therapeutic avenues for CLL.
慢性淋巴细胞白血病(CLL)是西方世界最常见的B细胞恶性肿瘤,其特征是尽管有暂时缓解,但仍频繁复发。我们的研究整合了公开可用的蛋白质组学、转录组学和患者生存数据集,以确定健康样本和CLL样本之间的关键差异。我们发现了大约1000种可区分健康细胞和癌细胞的蛋白质,其中608种在CLL病例中上调,415种下调。显著上调的蛋白质包括YEATS2(一种表观遗传调节因子)、PIGR(聚合免疫球蛋白受体)和SNRPA(一种剪接因子),它们可能作为这种疾病的预后生物标志物。与CLL进展相关的关键途径涉及RNA加工、应激抗性和免疫反应缺陷。此外,我们确定了三种现有药物——博舒替尼、伏立诺他和帕比司他——用于在CLL药物重新利用方面进行潜在的进一步研究。我们还发现转录组学和蛋白质组学数据之间的相关性有限,强调了蛋白质组学在理解基因表达调控机制方面的重要性。这种普遍已知的差异再次凸显,由于许多调控因素,如蛋白质降解、转录后修饰和不同的翻译速率,mRNA水平不能准确预测蛋白质丰度。这些结果证明了整合组学数据以揭示癌症中失调的蛋白质和途径的价值,并为CLL提出了新的治疗途径。