Beijing Life Science Academy, Beijing, China.
State Key Laboratory of Genetic Engineering, Department of Biochemistry, Collaborative Innovation Center of Genetics and Development, School of Life Sciences, Institute of Plant Biology, Fudan University, Shanghai 200438, China.
Mol Biol Evol. 2024 Sep 4;41(9). doi: 10.1093/molbev/msae178.
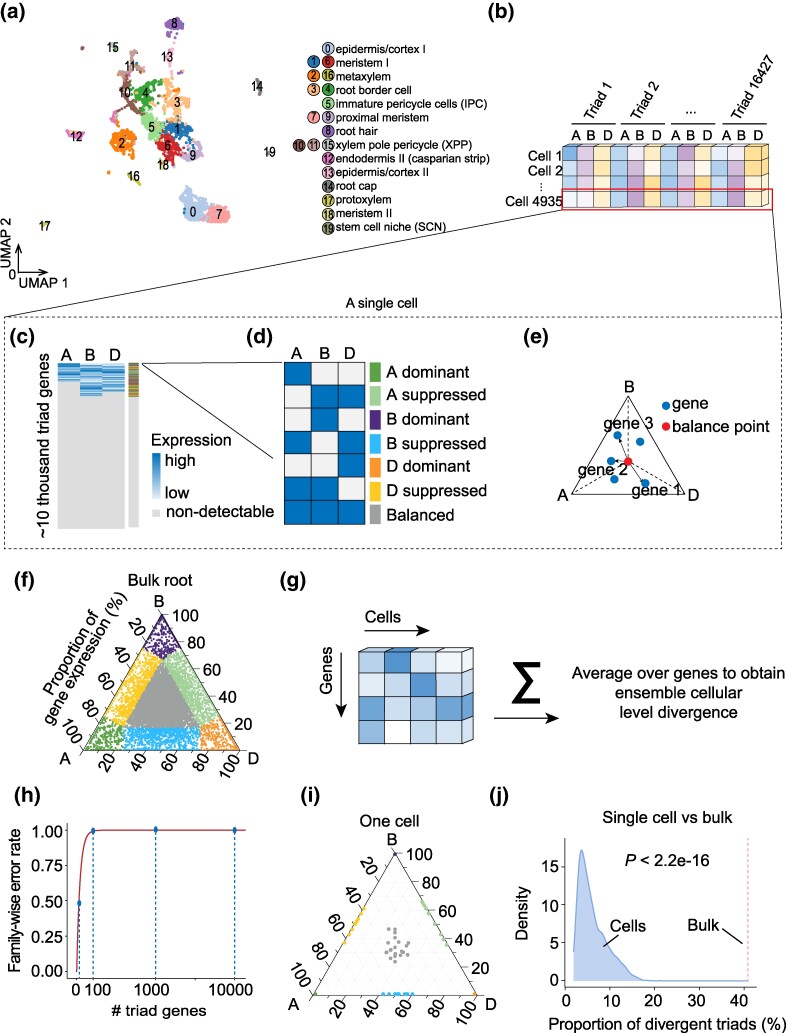
Polyploidization drives regulatory and phenotypic innovation. How the merger of different genomes contributes to polyploid development is a fundamental issue in evolutionary developmental biology and breeding research. Clarifying this issue is challenging because of genome complexity and the difficulty in tracking stochastic subgenome divergence during development. Recent single-cell sequencing techniques enabled probing subgenome-divergent regulation in the context of cellular differentiation. However, analyzing single-cell data suffers from high error rates due to high dimensionality, noise, and sparsity, and the errors stack up in polyploid analysis due to the increased dimensionality of comparisons between subgenomes of each cell, hindering deeper mechanistic understandings. In this study, we develop a quantitative computational framework, called "pseudo-genome divergence quantification" (pgDQ), for quantifying and tracking subgenome divergence directly at the cellular level. Further comparing with cellular differentiation trajectories derived from single-cell RNA sequencing data allows for an examination of the relationship between subgenome divergence and the progression of development. pgDQ produces robust results and is insensitive to data dropout and noise, avoiding high error rates due to multiple comparisons of genes, cells, and subgenomes. A statistical diagnostic approach is proposed to identify genes that are central to subgenome divergence during development, which facilitates the integration of different data modalities, enabling the identification of factors and pathways that mediate subgenome-divergent activity during development. Case studies have demonstrated that applying pgDQ to single-cell and bulk tissue transcriptomic data promotes a systematic and deeper understanding of how dynamic subgenome divergence contributes to developmental trajectories in polyploid evolution.
多倍化驱动了调控和表型的创新。不同基因组的融合如何促进多倍体的发育,这是进化发育生物学和育种研究中的一个基本问题。由于基因组的复杂性以及在发育过程中追踪随机亚基因组分化的困难,澄清这个问题具有挑战性。最近的单细胞测序技术使我们能够在细胞分化的背景下探测亚基因组分化的调控。然而,由于高维度、噪声和稀疏性,单细胞数据分析会受到高错误率的影响,而且由于每个细胞的亚基因组之间的比较维度增加,错误会在多倍体分析中累积,从而阻碍了更深入的机制理解。在这项研究中,我们开发了一种定量计算框架,称为“伪基因组分歧量化”(pgDQ),用于在细胞水平上直接量化和跟踪亚基因组分歧。进一步与单细胞 RNA 测序数据得出的细胞分化轨迹进行比较,使我们能够检查亚基因组分歧与发育进程之间的关系。pgDQ 产生了稳健的结果,并且对数据缺失和噪声不敏感,避免了由于基因、细胞和亚基因组的多次比较而导致的高错误率。提出了一种统计诊断方法来识别在发育过程中亚基因组分歧的关键基因,这有助于整合不同的数据模态,使我们能够识别在发育过程中介导亚基因组分化活性的因素和途径。案例研究表明,将 pgDQ 应用于单细胞和批量组织转录组数据可以促进系统和更深入地理解动态的亚基因组分歧如何促进多倍体进化中的发育轨迹。