Department of Neurology and Stroke Medicine, Yokohama City University Graduate School of Medicine, 3-9 Fukuura, Kanazawa-Ku, Yokohama, 236-0004, Japan.
Department of Human Genetics, Yokohama City University Graduate School of Medicine, 3-9 Fukuura, Kanazawa-Ku, Yokohama, 236-0004, Japan.
BMC Neurol. 2024 Sep 4;24(1):310. doi: 10.1186/s12883-024-03823-9.
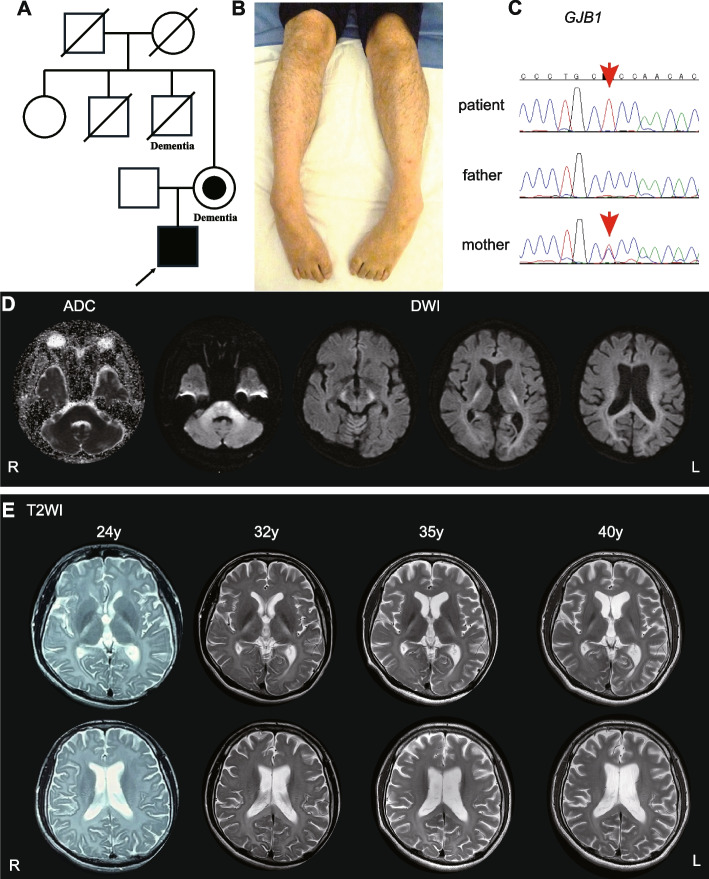
Pathogenic variants in Gap junction protein beta 1 (GJB1), which encodes Connexin 32, are known to cause X-linked Charcot-Marie-Tooth disease (CMTX), the second most common form of CMT. CMTX presents with the following five central nervous systems (CNS) phenotypes: subclinical electrophysiological abnormalities, mild fixed abnormalities on neurological examination and/or imaging, transient CNS dysfunction, cognitive impairment, and persistent CNS manifestations.
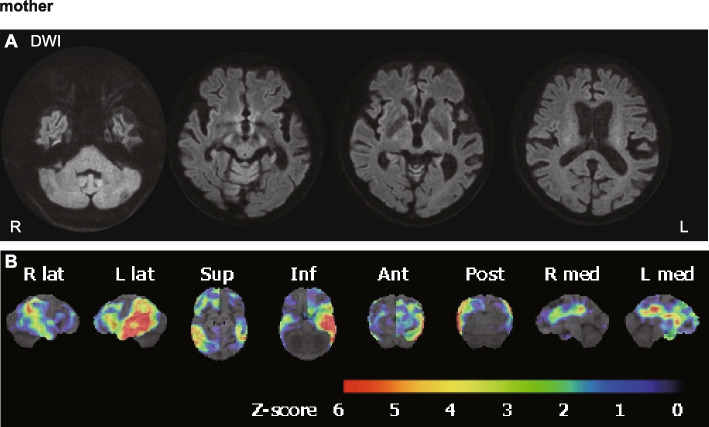
A 40-year-old Japanese male showed CNS symptoms, including nystagmus, prominent spastic paraplegia, and mild cerebellar ataxia, accompanied by subclinical peripheral neuropathy. Brain magnetic resonance imaging revealed hyperintensities in diffusion-weighted images of the white matter, particularly along the pyramidal tract, which had persisted since childhood. Nerve conduction assessment showed a mild decrease in motor conduction velocity, and auditory brainstem responses beyond wave II were absent. Peripheral and central conduction times in somatosensory evoked potentials elicited by stimulation of the median nerve were prolonged. Genetic analysis identified a hemizygous GJB1 variant, NM_000166.6:c.520C > T p.Pro174Ser.
The patient in the case described here, with a GJB1 p.Pro174Ser variant, presented with a unique CNS-dominant phenotype, characterized by spastic paraplegia and persistent extensive leukoencephalopathy, rather than CMTX. Similar phenotypes have also been observed in patients with GJC2 and CLCN2 variants, likely because of the common function of these genes in regulating ion and water balance, which is essential for maintaining white matter function. CMTX should be considered within the spectrum of GJB1-related disorders, which can include patients with predominant CNS symptoms, some of which can potentially be classified as a new type of spastic paraplegia.
Gap junction protein beta 1(GJB1)基因中的致病性变异可导致 X 连锁遗传性运动感觉神经病(CMTX),这是 CMT 的第二大常见类型。CMTX 存在以下五种中枢神经系统(CNS)表型:亚临床电生理学异常、神经检查和/或影像学上轻度固定异常、短暂性 CNS 功能障碍、认知障碍和持续性 CNS 表现。
一名 40 岁的日本男性出现 CNS 症状,包括眼球震颤、显著痉挛性截瘫和轻度小脑共济失调,伴有亚临床周围神经病。脑磁共振成像显示白质弥散加权图像中存在高信号,特别是沿锥体束,自儿童期以来一直存在。神经传导评估显示运动传导速度轻度降低,听觉脑干反应中 II 波以后缺失。刺激正中神经诱发的体感诱发电位的外周和中枢传导时间延长。基因分析发现 GJB1 基因 NM_000166.6:c.520C>T p.Pro174Ser 半合子变异。
本病例中患者携带 GJB1 p.Pro174Ser 变异,表现出独特的以 CNS 为主的表型,特征为痉挛性截瘫和持续性广泛脑白质病变,而不是 CMTX。GJC2 和 CLCN2 变异的患者也观察到类似的表型,可能是因为这些基因的共同功能是调节离子和水的平衡,这对于维持白质功能至关重要。CMTX 应被认为是 GJB1 相关疾病谱的一部分,其中可能包括以 CNS 症状为主的患者,其中一些可能潜在地被归类为新型痉挛性截瘫。