Department of Neuromuscular Diseases, UCL Queen Square Institute of Neurology, London, WC1N 3BG, UK.
Department of Clinical Neurosciences, Fondazione IRCCS Istituto Neurologico Carlo Besta, 20133 Milan, Italy.
Brain. 2023 Oct 3;146(10):4336-4349. doi: 10.1093/brain/awad187.
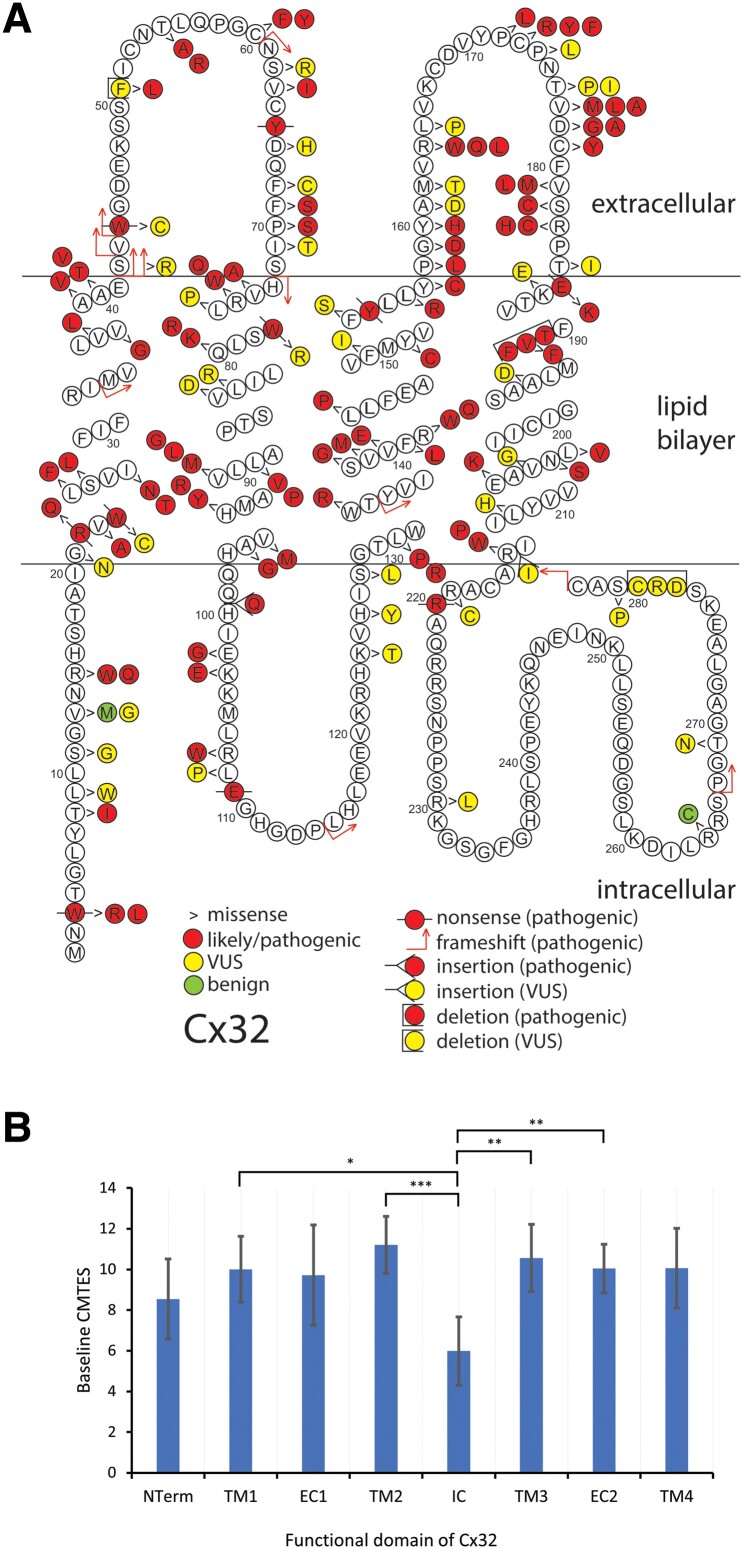
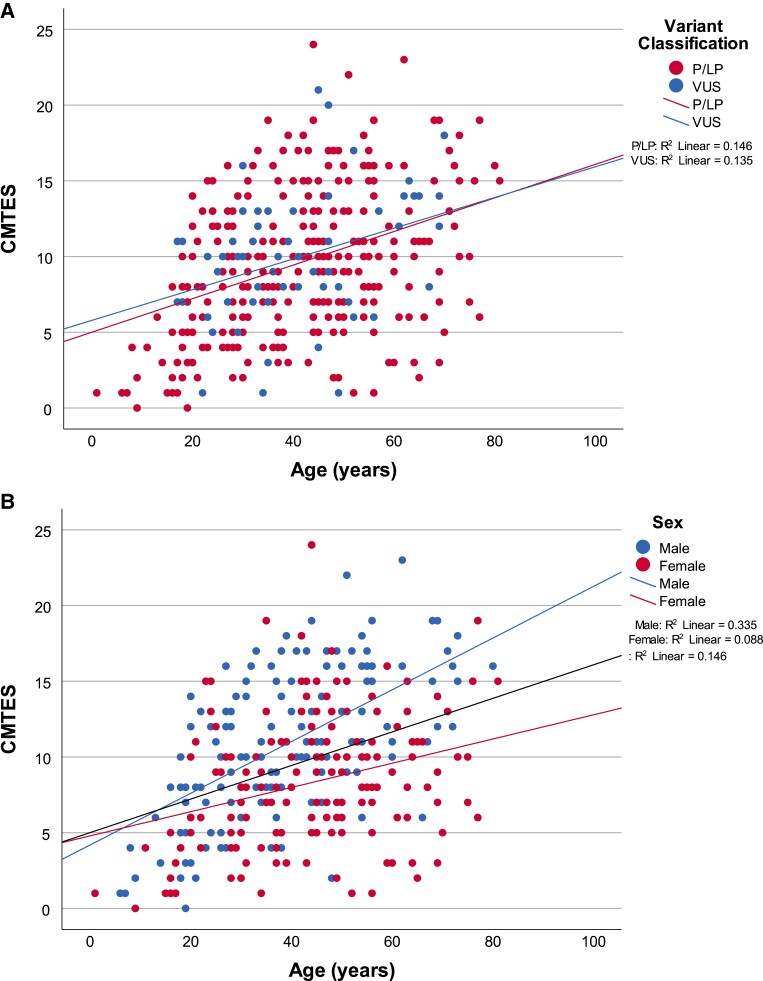
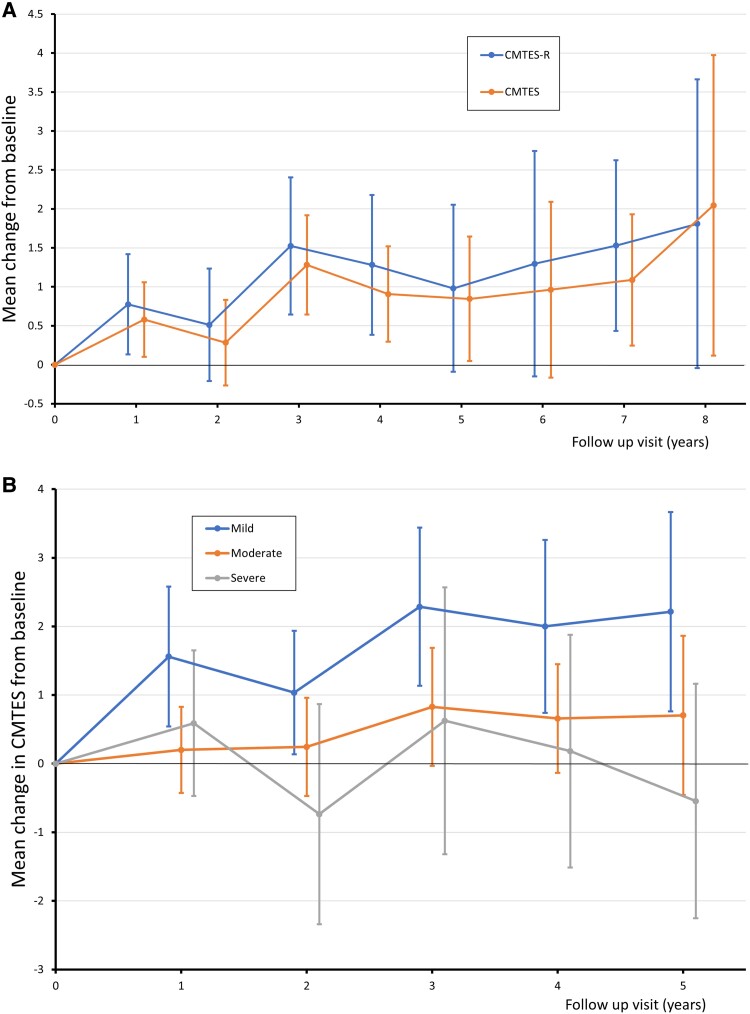
Charcot-Marie-Tooth disease (CMT) due to GJB1 variants (CMTX1) is the second most common form of CMT. It is an X-linked disorder characterized by progressive sensory and motor neuropathy with males affected more severely than females. Many reported GJB1 variants remain classified as variants of uncertain significance (VUS). In this large, international, multicentre study we prospectively collected demographic, clinical and genetic data on patients with CMT associated with GJB1 variants. Pathogenicity for each variant was defined using adapted American College of Medical Genetics criteria. Baseline and longitudinal analyses were conducted to study genotype-phenotype correlations, to calculate longitudinal change using the CMT Examination Score (CMTES), to compare males versus females, and pathogenic/likely pathogenic (P/LP) variants versus VUS. We present 387 patients from 295 families harbouring 154 variants in GJB1. Of these, 319 patients (82.4%) were deemed to have P/LP variants, 65 had VUS (16.8%) and three benign variants (0.8%; excluded from analysis); an increased proportion of patients with P/LP variants compared with using ClinVar's classification (74.6%). Male patients (166/319, 52.0%, P/LP only) were more severely affected at baseline. Baseline measures in patients with P/LP variants and VUS showed no significant differences, and regression analysis suggested the disease groups were near identical at baseline. Genotype-phenotype analysis suggested c.-17G>A produces the most severe phenotype of the five most common variants, and missense variants in the intracellular domain are less severe than other domains. Progression of disease was seen with increasing CMTES over time up to 8 years follow-up. Standard response mean (SRM), a measure of outcome responsiveness, peaked at 3 years with moderate responsiveness [change in CMTES (ΔCMTES) = 1.3 ± 2.6, P = 0.00016, SRM = 0.50]. Males and females progressed similarly up to 8 years, but baseline regression analysis suggested that over a longer period, females progress more slowly. Progression was most pronounced for mild phenotypes (CMTES = 0-7; 3-year ΔCMTES = 2.3 ± 2.5, P = 0.001, SRM = 0.90). Enhanced variant interpretation has yielded an increased proportion of GJB1 variants classified as P/LP and will aid future variant interpretation in this gene. Baseline and longitudinal analysis of this large cohort of CMTX1 patients describes the natural history of the disease including the rate of progression; CMTES showed moderate responsiveness for the whole group at 3 years and higher responsiveness for the mild group at 3, 4 and 5 years. These results have implications for patient selection for upcoming clinical trials.
Charcot-Marie-Tooth 病(CMT)由于 GJB1 变体(CMTX1)是 CMT 的第二大常见形式。它是一种 X 连锁疾病,其特征是进行性感觉和运动神经病,男性比女性受影响更严重。许多报道的 GJB1 变体仍然被归类为意义不明的变体(VUS)。在这项大型国际多中心研究中,我们前瞻性地收集了与 GJB1 变体相关的 CMT 患者的人口统计学、临床和遗传数据。使用改编的美国医学遗传学学院标准来定义每个变体的致病性。进行了基线和纵向分析,以研究基因型-表型相关性,使用 CMT 检查评分(CMTES)计算纵向变化,比较男性与女性,以及致病性/可能致病性(P/LP)变体与 VUS。我们展示了来自 295 个家族的 387 名携带 GJB1 中 154 种变体的患者。其中,319 名患者(82.4%)被认为携带 P/LP 变体,65 名患者携带 VUS(16.8%),3 名患者携带良性变体(0.8%;排除在分析之外);与使用 ClinVar 的分类相比,具有 P/LP 变体的患者比例增加(74.6%)。基线时,仅携带 P/LP 变体的男性患者(166/319,52.0%)受影响更严重。携带 P/LP 变体和 VUS 的患者的基线测量值没有显著差异,回归分析表明,在基线时,疾病组几乎相同。基因型-表型分析表明,c.-17G>A 产生了五个最常见变体中最严重的表型,细胞内结构域的错义变体比其他结构域的变体更不严重。随着时间的推移,CMTES 增加,疾病进展可见,随访 8 年。标准反应均值(SRM)是一种衡量结果反应性的指标,在 3 年时达到峰值,具有中度反应性[CMTES 的变化(ΔCMTES)= 1.3 ± 2.6,P = 0.00016,SRM = 0.50]。男性和女性在 8 年内进展相似,但基线回归分析表明,在较长时间内,女性进展较慢。进展最明显的是轻度表型(CMTES=0-7;3 年ΔCMTES=2.3±2.5,P=0.001,SRM=0.90)。对变体的解释增强产生了更高比例的 GJB1 变体被归类为 P/LP,并将有助于未来在该基因中进行变体解释。对大量 CMTX1 患者的基线和纵向分析描述了疾病的自然史,包括进展速度;CMTES 在 3 年时对整个组具有中度反应性,在 3、4 和 5 年时对轻度组具有更高的反应性。这些结果对即将进行的临床试验中的患者选择具有影响。