Liu Liang, Yu Lili, Wu Shaoyuan, Arnold Jonathan, Whalen Christopher, Davis Charles, Edwards Scott
Department of Statistics and Institute of Bioinformatics, University of Georgia, Athens, GA, United States.
Department of Biostatistics, Georgia Southern University, Statesboro, GA, United States.
Front Ecol Evol. 2023;11. doi: 10.3389/fevo.2023.1134764. Epub 2023 Jun 28.
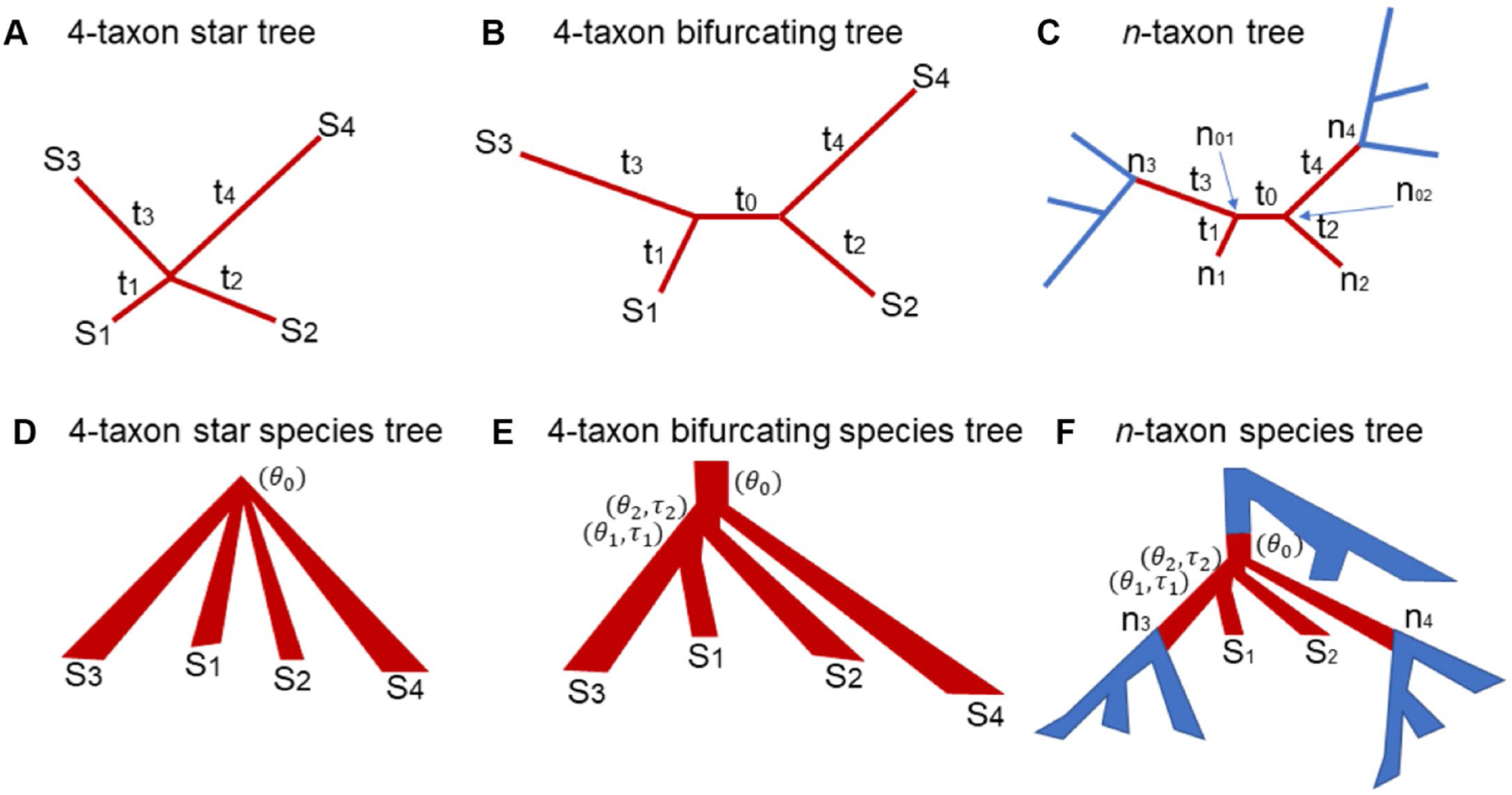
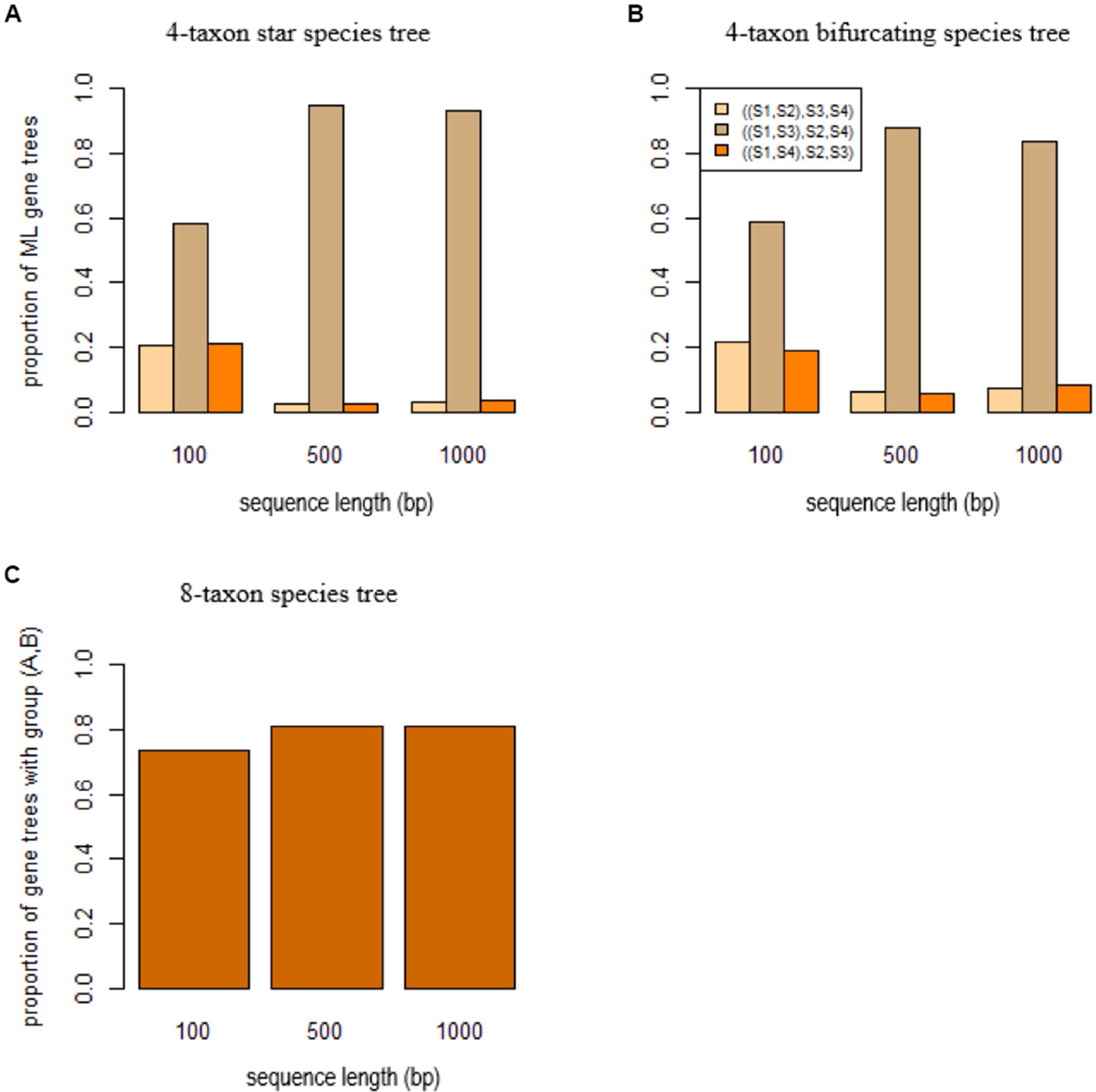
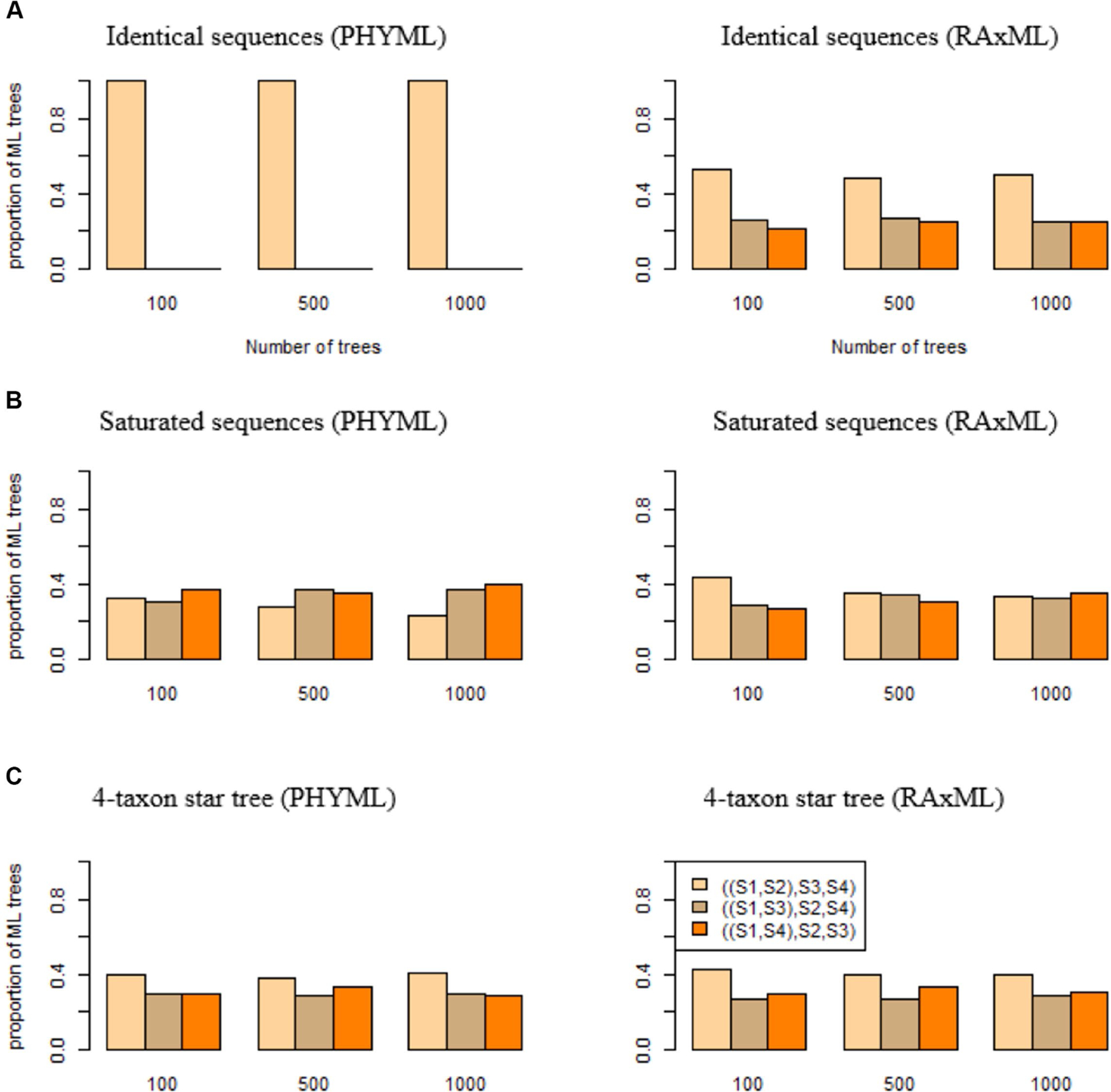
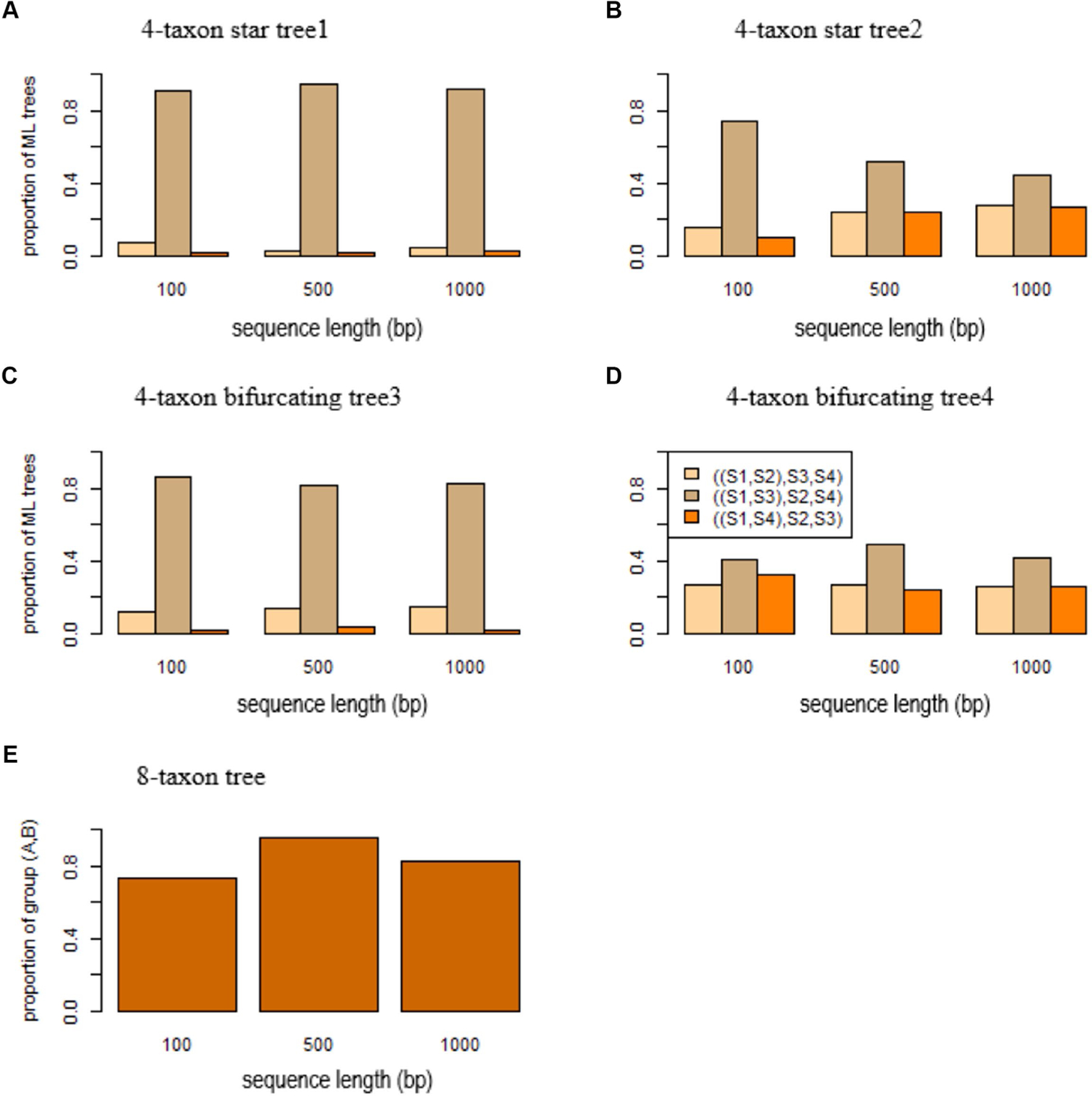
Accurate reconstruction of species trees often relies on the quality of input gene trees estimated from molecular sequences. Previous studies suggested that if the sequence length is fixed, the maximum likelihood may produce biased gene trees which subsequently mislead inference of species trees. Two key questions need to be answered in this context: what are the scenarios that may result in consistently biased gene trees? and for those scenarios, are there any remedies that may remove or at least reduce the misleading effects of consistently biased gene trees? In this article, we establish a theoretical framework to address these questions. Considering a scenario where the true gene tree is a 4-taxon star tree with two short branches leading to the species and , we demonstrate that maximum likelihood significantly favors the wrong bifurcating tree grouping the two species and with short branches. We name this inconsistent behavior short branch attraction, which may occur in real-world data involving a 4-taxon bifurcating gene tree with a short internal branch. If no mutation occurs along the internal branch, which is likely if the internal branch is short, the 4-taxon bifurcating tree is equivalent to the 4-taxon star tree and thus will suffer the same misleading effect of short branch attraction. Theoretical and simulation results further demonstrate that short branch attraction may occur in gene trees and species trees of arbitrary size. Moreover, short branch attraction is primarily caused by a lack of phylogenetic information in sequence data, suggesting that converting short internal branches to polytomies in the estimated gene trees can significantly reduce artifacts induced by short branch attraction.
物种树的准确重建通常依赖于从分子序列估计的输入基因树的质量。先前的研究表明,如果序列长度固定,最大似然法可能会产生有偏差的基因树,进而误导物种树的推断。在这种情况下,有两个关键问题需要回答:哪些情况可能导致基因树持续出现偏差?对于这些情况,是否有任何补救措施可以消除或至少减少持续有偏差的基因树的误导性影响?在本文中,我们建立了一个理论框架来解决这些问题。考虑到真实基因树是一个具有四条分类单元的星状树,有两条短分支通向物种 和 的情况,我们证明最大似然法显著倾向于错误的二叉树,即将两个具有短分支的物种 和 归为一组。我们将这种不一致的行为称为短分支吸引,它可能出现在涉及具有短内部分支的四条分类单元二叉基因树的实际数据中。如果沿着内部分支没有发生突变(如果内部分支很短,这种情况很可能发生),那么四条分类单元二叉树就等同于四条分类单元星状树,因此会受到相同的短分支吸引的误导性影响。理论和模拟结果进一步表明,短分支吸引可能出现在任意大小的基因树和物种树中。此外,短分支吸引主要是由序列数据中缺乏系统发育信息引起的,这表明在估计的基因树中将短内部分支转换为多歧分支可以显著减少由短分支吸引引起的假象。