Guerrini-Rousseau Lea, Merlevede Jane, Denizeau Philippe, Andreiuolo Felipe, Varlet Pascale, Puget Stéphanie, Beccaria Kevin, Blauwblomme Thomas, Cabaret Odile, Hamzaoui Nadim, Bourdeaut Franck, Faure-Conter Cécile, Muleris Martine, Colas Chrystelle, Adam de Beaumais Tiphaine, Castel David, Rouleau Etienne, Brugières Laurence, Grill Jacques, Debily Marie-Anne
Department of Children and Adolescents Oncology, Gustave Roussy, Villejuif, France.
Molecular Predictors and New Targets in Oncology, INSERM U981, Team "Genomics and Oncogenesis of pediatric Brain Tumors," Gustave Roussy, Université Paris-Saclay, Villejuif, France.
Neurooncol Adv. 2024 Jul 11;6(1):vdae120. doi: 10.1093/noajnl/vdae120. eCollection 2024 Jan-Dec.
Constitutional mismatch repair deficiency (CMMRD) is a cancer predisposition due to biallelic mutations in one of the mismatch repair (MMR) genes associated with early onset of cancers, especially high-grade gliomas. Our aim was to decipher the molecular specificities of these gliomas.
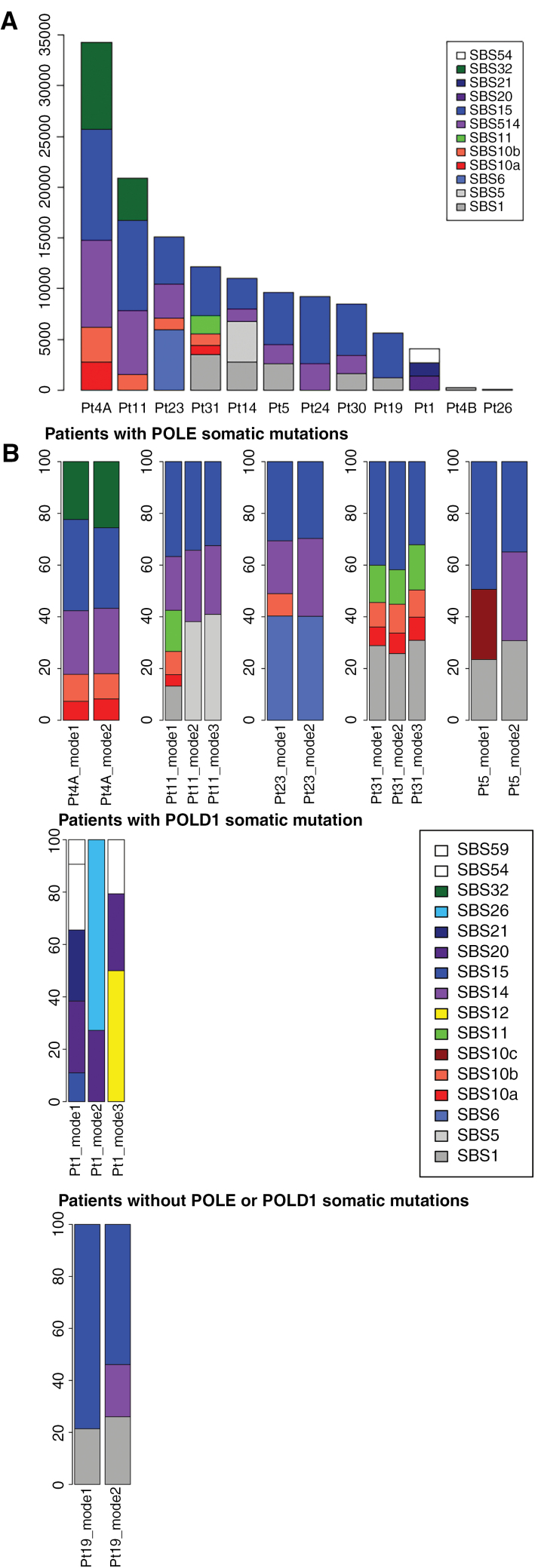
Clinical, histopathological, and whole exome sequencing data were analyzed in 12 children with genetically proven CMMRD and a high-grade glioma.
PDL1 expression was present in immunohistochemistry in 50% of the samples. In 9 patients, the glioma harbored an ultra-hypermutated phenotype (104-635 coding single nucleotide variants (SNV) per Mb, median 204). Driver mutations in and exonuclease domains were described for 8 and 1 patients respectively and were always present in the mutation burst with the highest variant allele frequency (VAF). The mutational signatures were dominated by MMR-related ones and similar in the different mutation bursts of a same patient without subsequent enrichment of the mutation signatures with POL-driven ones. Median number of coding SNV with VAF above one of the driving polymerase mutation per Mb was 57 (17-191). Our findings suggest that somatic polymerase alterations does not entirely explain the ultra-hypermutant phenotype. , , , , and genes were frequently mutated with higher VAF than the deleterious somatic polymerase mutation.
CMMRD-associated gliomas have a specific oncogenesis that does not involve usual pathways and mutations seen in sporadic pediatric or adult glioblastomas. Frequent alterations in other pathways such as MAPK may suggest the use of other targeted therapies along with PD1 inhibitors.
遗传性错配修复缺陷(CMMRD)是一种癌症易感性疾病,由错配修复(MMR)基因之一的双等位基因突变引起,与癌症的早期发生有关,尤其是高级别胶质瘤。我们的目的是解读这些胶质瘤的分子特异性。
对12例经基因证实患有CMMRD且患有高级别胶质瘤的儿童的临床、组织病理学和全外显子测序数据进行了分析。
免疫组织化学检测显示50%的样本中存在程序性死亡受体1(PDL1)表达。在9例患者中,胶质瘤具有超高度突变表型(每兆碱基104 - 635个编码单核苷酸变异(SNV),中位数为204)。分别在8例和1例患者中描述了核酸外切酶结构域的驱动突变,且这些突变总是出现在具有最高变异等位基因频率(VAF)的突变爆发中。突变特征以与MMR相关的特征为主,在同一患者的不同突变爆发中相似,后续未出现由DNA聚合酶(POL)驱动的突变特征富集。每兆碱基中VAF高于驱动性聚合酶突变之一的编码SNV中位数为57(17 - 191)。我们的研究结果表明,体细胞聚合酶改变并不能完全解释超高度突变表型。表皮生长因子受体(EGFR)、神经纤维瘤病1型(NF1)、磷酸肌醇-3-激酶调节亚基1(PIK3R1)、肿瘤蛋白p53(TP53)、磷酸酶及张力蛋白同源物(PTEN)和神经母细胞瘤RAS病毒癌基因同源物(NRAS)基因经常发生突变,其VAF高于有害的体细胞聚合酶突变。
CMMRD相关的胶质瘤具有特定的肿瘤发生机制,不涉及散发性儿童或成人胶质母细胞瘤中常见的途径和突变。丝裂原活化蛋白激酶(MAPK)等其他途径的频繁改变可能提示除了程序性死亡受体1(PD1)抑制剂外,还可使用其他靶向治疗。