Ahdi Saher Gul, Alvi Javeria Raza, Ashfaq Azeem, Sultan Tipu
Dr. Saher Gul Ahdi University of Child Health Sciences, The Children's Hospital, Lahore, Pakistan.
Dr. Javeria Raza Alvi University of Child Health Sciences, The Children's Hospital, Lahore, Pakistan.
Pak J Med Sci. 2024 Sep;40(8):1638-1643. doi: 10.12669/pjms.40.8.8006.
To unravel the clinical and genetic specifications of Neuronal ceroid lipofuscinosis (NCL).
This is a retrospective cross-sectional study conducted in the Department of Pediatric Neurology Children Hospital and University of Child Health Sciences, Lahore, Pakistan from March 2017 to March 2022. The primary outcome was to measure genotype-phenotype correlation by segregation of phenotypes according to genotype. The secondary outcomes included a correlation between genotype and distribution of age(s) of onset.
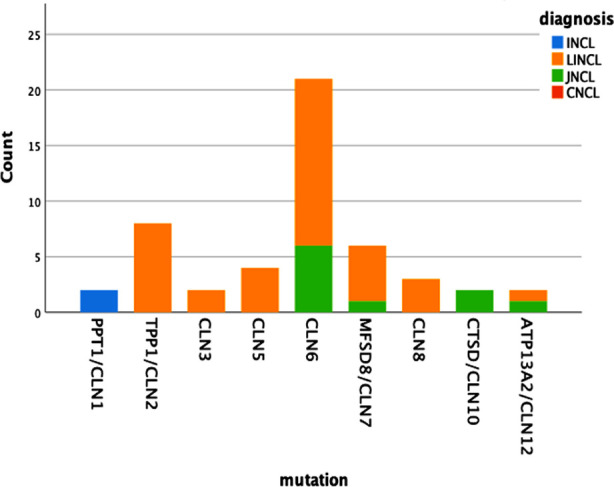
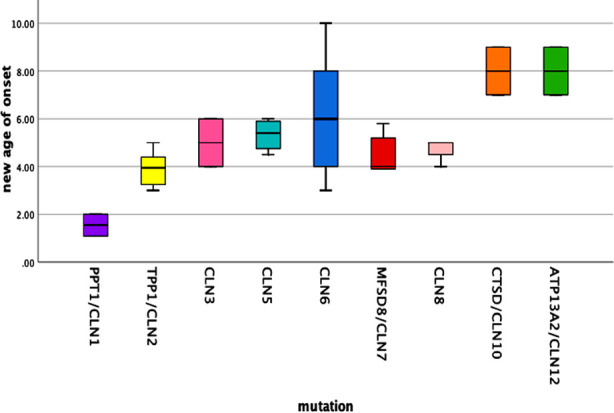
One hundred fifty three patients clinically diagnosed with NCL underwent genetic testing and pathologic mutation was identified in 32.7% of patients. About 59.6% were male and 37.2% had an affected sibling. The median age was 5.46±1.95 years at the onset of the first symptom i.e., myoclonic seizures in 68%, and motor difficulty in 24%. Other features found were global developmental delay (56%), hypotonia (23%), visual impairment (80%), ataxia (22%), and disc pallor (56%). The most common type was CLN6 (Ceroid lipofuscinosis neuronal) (42%), CLN2 (16%) followed by CLN7 (12%). When 50 patients with recognized mutations were compared with 103 patients with no mutation, family history (p=0.049), early visual loss (p=0.016), hypotonia (p=0.001), white matter signals (p=0.026) and pan-atrophy(p=0.047) was statistically significant in the genetically confirmed NCL. Multiple pairwise comparisons indicated that the estimated age of onset for the CLN1 and CLN2 mutation group was significantly lower than other genotypes including CLN6 (p 0.012), CLN10 (p 0.007) and CLN12 (p 0.007).
Following a detailed review of NCL symptomatology, a clinically-oriented approach should be used for a rapid diagnosis with confirmation by targeted molecular testing for future genetic counseling.
揭示神经元蜡样脂褐质沉积症(NCL)的临床和遗传特征。
这是一项回顾性横断面研究,于2017年3月至2022年3月在巴基斯坦拉合尔儿童医院和儿童健康科学大学儿科神经科进行。主要结果是根据基因型对表型进行分类,以测量基因型与表型的相关性。次要结果包括基因型与发病年龄分布之间的相关性。
153例临床诊断为NCL的患者接受了基因检测,32.7%的患者检测到病理性突变。约59.6%为男性,37.2%有患病同胞。首次出现症状时的中位年龄为5.46±1.95岁,其中68%为肌阵挛性癫痫发作,24%为运动困难。其他发现的特征包括全面发育迟缓(56%)、肌张力减退(23%)、视力损害(80%)、共济失调(22%)和视盘苍白(56%)。最常见的类型是CLN6(神经元蜡样脂褐质沉积症)(42%),CLN2(16%),其次是CLN7(12%)。当将50例有已知突变的患者与103例无突变的患者进行比较时,家族史(p=0.049)、早期视力丧失(p=0.016)、肌张力减退(p=0.001)、白质信号(p=0.026)和全身萎缩(p=0.047)在基因确诊的NCL中具有统计学意义。多重成对比较表明,CLN1和CLN2突变组的估计发病年龄显著低于其他基因型,包括CLN6(p<0.012)、CLN10(p<0.007)和CLN12(p<0.007)。
在对NCL症状学进行详细回顾后,应采用以临床为导向的方法进行快速诊断,并通过靶向分子检测进行确认,以便未来进行遗传咨询。