Department of Biotechnology and Genetic Engineering, Kohat University of Science and Technology, Kohat, 26000, Khyber Pakhtunkhwa, Pakistan.
2Medical Research, RILD Wellcome Wolfson Centre (Level 4), Royal Devon and Exeter NHS Foundation Trust, Exeter, Devon, EX2 5DW, UK.
BMC Neurol. 2024 Sep 20;24(1):354. doi: 10.1186/s12883-024-03855-1.
Hereditary Spastic Paraplegias (HSPs) and Hereditary Cerebellar Ataxias (HCAs) are progressive neurodegenerative disorders encompassing a spectrum of neurogenetic conditions with significant overlaps of clinical features. Spastic ataxias are a group of conditions that have features of both cerebellar ataxia and spasticity, and these conditions are frequently clinically challenging to distinguish. Accurate genetic diagnosis is crucial but challenging, particularly in resource-limited settings. This study aims to investigate the genetic basis of HSPs and HCAs in Pakistani families.
Families from Khyber Pakhtunkhwa with at least two members showing HSP or HCA phenotypes, and who had not previously been analyzed genetically, were included. Families were referred for genetic analysis by local neurologists based on the proband's clinical features and signs of a potential genetic neurodegenerative disorder. Whole Exome Sequencing (WES) and Sanger sequencing were then used to identify and validate genetic variants, and to analyze variant segregation within families to determine inheritance patterns. The mean age of onset and standard deviation were calculated to assess variability among affected individuals, and the success rate was compared with literature reports using differences in proportions and Cohen's h.
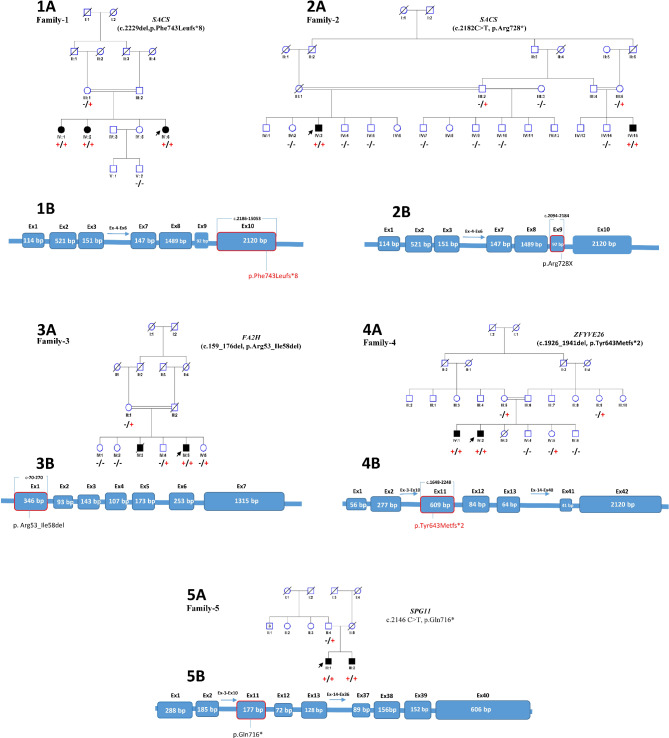
Pathogenic variants associated with these conditions were identified in five of eight families, segregating according to autosomal recessive inheritance. These variants included previously reported SACS c.2182 C > T, p.(Arg728*), FA2H c.159_176del, p.(Arg53_Ile58del) and SPG11 c.2146 C > T, p.(Gln716*) variants, and two previously unreported variants in SACS c.2229del, p.(Phe743Leufs8) and ZFYVE26 c.1926_1941del, p.(Tyr643Metfs2). Additionally, FA2H and SPG11 variants were found to have recurrent occurrences, suggesting a potential founder effect within the Pakistani population. Onset age among affected individuals ranged from 1 to 14 years (M = 6.23, SD = 3.96). The diagnostic success rate was 62.5%, with moderate effect sizes compared to previous studies.
The findings of this study expand the genotypic and phenotypic spectrum of HSPs and HCAs in Pakistan and emphasize the importance of utilizing exome/genome sequencing for accurate diagnosis or support accurate differential diagnosis. This approach can improve genetic counseling and clinical management, addressing the challenges of diagnosing neurodegenerative disorders in resource-limited settings.
遗传性痉挛性截瘫(HSPs)和遗传性小脑共济失调(HCAs)是进行性神经退行性疾病,涵盖了一系列具有显著临床特征重叠的神经遗传疾病。痉挛性共济失调是一组具有小脑共济失调和痉挛特征的疾病,这些疾病在临床上经常难以区分。准确的基因诊断至关重要,但具有挑战性,特别是在资源有限的环境中。本研究旨在探讨巴基斯坦家族中 HSPs 和 HCAs 的遗传基础。
本研究纳入了来自开伯尔-普赫图赫瓦省(Khyber Pakhtunkhwa)的至少有两名表现出 HSP 或 HCA 表型的家庭成员的家族,这些家族之前没有进行过基因分析。当地神经科医生根据先证者的临床特征和潜在遗传神经退行性疾病的迹象,将这些家族转介进行基因分析。然后使用全外显子组测序(WES)和 Sanger 测序来识别和验证遗传变异,并分析家系内变异的分离,以确定遗传模式。计算发病年龄的平均值和标准差,以评估受影响个体之间的变异性,并使用比例差异和 Cohen's h 与文献报告进行成功率比较。
在 8 个家族中的 5 个家族中发现了与这些疾病相关的致病性变异,这些变异符合常染色体隐性遗传模式。这些变异包括先前报道的 SACS c.2182C>T,p.(Arg728*)、FA2H c.159_176del,p.(Arg53_Ile58del)和 SPG11 c.2146C>T,p.(Gln716*)变异,以及 SACS c.2229del,p.(Phe743Leufs8)和 ZFYVE26 c.1926_1941del,p.(Tyr643Metfs2)这两个以前未报道的变异。此外,还发现 FA2H 和 SPG11 变异具有复发性,提示巴基斯坦人群中存在潜在的创始人效应。受影响个体的发病年龄范围为 1 至 14 岁(M=6.23,SD=3.96)。诊断成功率为 62.5%,与之前的研究相比,具有中等效应量。
本研究的结果扩展了 HSPs 和 HCAs 在巴基斯坦的基因型和表型谱,并强调了利用外显子/基因组测序进行准确诊断或支持准确鉴别诊断的重要性。这种方法可以改善遗传咨询和临床管理,解决资源有限环境中诊断神经退行性疾病的挑战。