Schillinger Jasmin, Koci Michelle, Bravo-Rodriguez Kenny, Heilmann Geronimo, Kaschani Farnusch, Kaiser Markus, Beuck Christine, Luecke Hartmut, Huber Robert, Hellerschmied Doris, Burston Steven G, Ehrmann Michael
Center of Medical Biotechnology, Faculty of Biology, University Duisburg-Essen, Essen, Germany.
Max-Planck-Institute of Molecular Physiology, Dortmund, Germany.
J Biol Chem. 2024 Nov;300(11):107812. doi: 10.1016/j.jbc.2024.107812. Epub 2024 Sep 21.
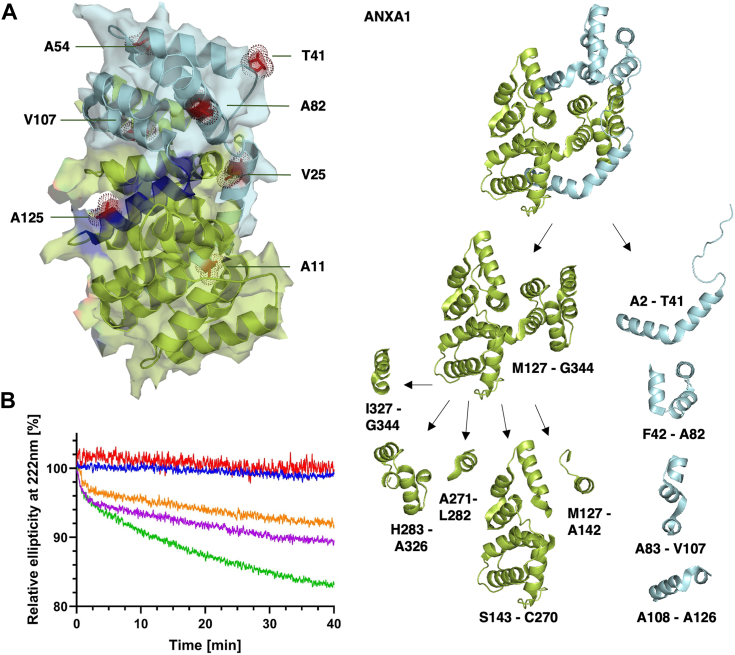
Members of the widely conserved high temperature requirement A (HtrA) family of serine proteases are involved in multiple aspects of protein quality control. In this context, they have been shown to efficiently degrade misfolded proteins or protein fragments. However, recent reports suggest that folded proteins can also be native substrates. To gain a deeper understanding of how folded proteins are initially processed and subsequently degraded into short peptides by human HTRA1, we established an integrated and quantitative approach using time-resolved mass spectrometry, CD spectroscopy, and bioinformatics. The resulting data provide high-resolution information on up to 178 individual proteolytic sites within folded ANXA1 (consisting of 346 amino acids), the relative frequency of cuts at each proteolytic site, the preferences of the protease for the amino acid sequence surrounding the scissile bond, as well as the degrees of sequential structural relaxation and unfolding of the substrate that occur during progressive degradation. Our workflow provides precise molecular insights into protease-substrate interactions, which could be readily adapted to address other posttranslational modifications such as phosphorylation in dynamic protein complexes.
丝氨酸蛋白酶的广泛保守的高温需求A(HtrA)家族成员参与蛋白质质量控制的多个方面。在这种情况下,它们已被证明能有效降解错误折叠的蛋白质或蛋白质片段。然而,最近的报道表明,折叠蛋白也可以是天然底物。为了更深入地了解折叠蛋白是如何最初被加工,随后被人HTRA1降解为短肽的,我们建立了一种综合定量方法,使用时间分辨质谱、圆二色光谱和生物信息学。所得数据提供了关于折叠的膜联蛋白A1(由346个氨基酸组成)内多达178个单个蛋白水解位点的高分辨率信息、每个蛋白水解位点的切割相对频率、蛋白酶对切割位点周围氨基酸序列的偏好,以及在渐进降解过程中底物发生的顺序结构松弛和解折叠程度。我们的工作流程为蛋白酶-底物相互作用提供了精确的分子见解,这可以很容易地用于研究其他翻译后修饰,如动态蛋白复合物中的磷酸化。