Department of Pediatrics, University of California, San Francisco, CA, USA.
Department of Nephrology, Alberta Children's Hospital, Calgary, AB, Canada.
Orphanet J Rare Dis. 2024 Sep 27;19(1):355. doi: 10.1186/s13023-024-03311-w.
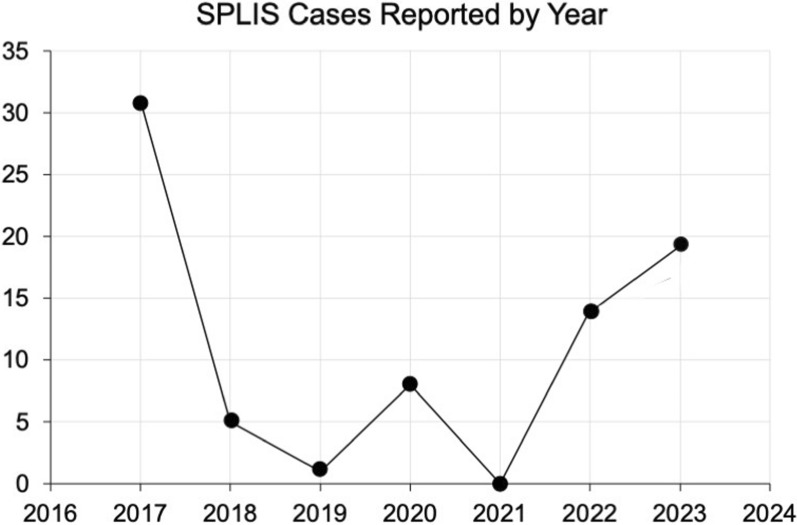
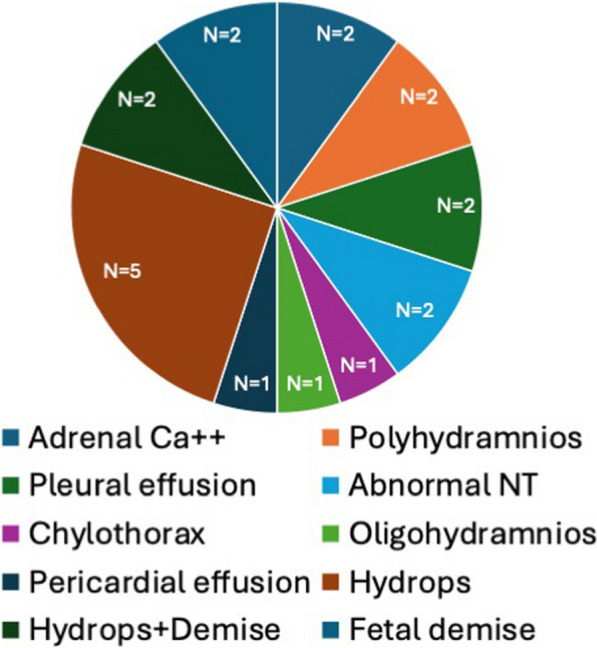
Sphingosine-1-phosphate lyase insufficiency syndrome (SPLIS) is a recently recognized inborn error of metabolism associated with steroid-resistant nephrotic syndrome as well as adrenal insufficiency and immunological, neurological, and skin manifestations. SPLIS is caused by inactivating mutations in SGPL1, encoding the pyridoxal 5'phosphate-dependent enzyme sphingosine-1-phosphate lyase, which catalyzes the final step of sphingolipid metabolism. Some SPLIS patients have undergone kidney transplantation, and others have been treated with vitamin B6 supplementation. In addition, targeted therapies including gene therapy are in preclinical development. In anticipation of clinical trials, it will be essential to characterize the full spectrum and natural history of SPLIS. We performed a retrospective analysis of 76 patients in whom the diagnosis of SPLIS was established in a proband with at least one suggestive finding and biallelic SGPL1 variants identified by molecular genetic testing. The main objective of the study was to identify factors influencing survival in SPLIS subjects.
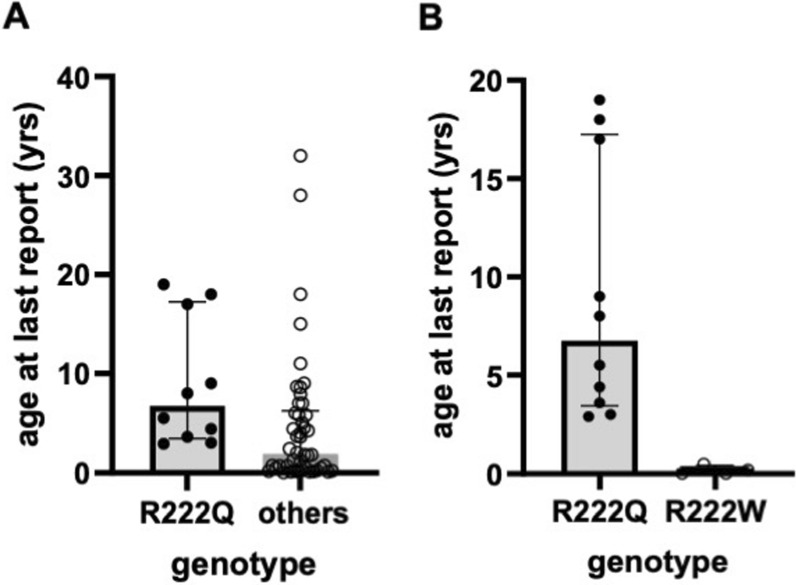
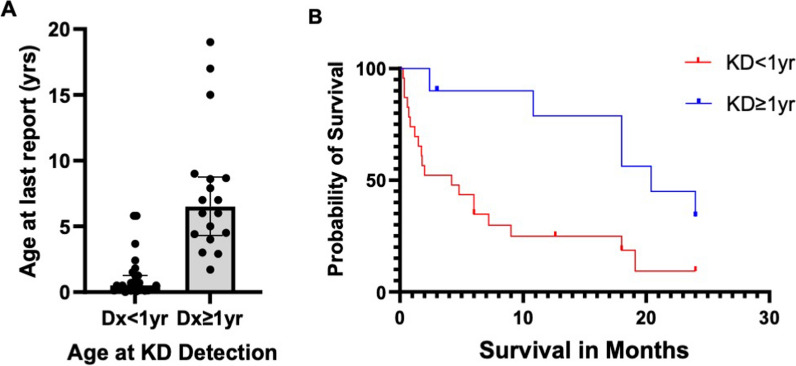
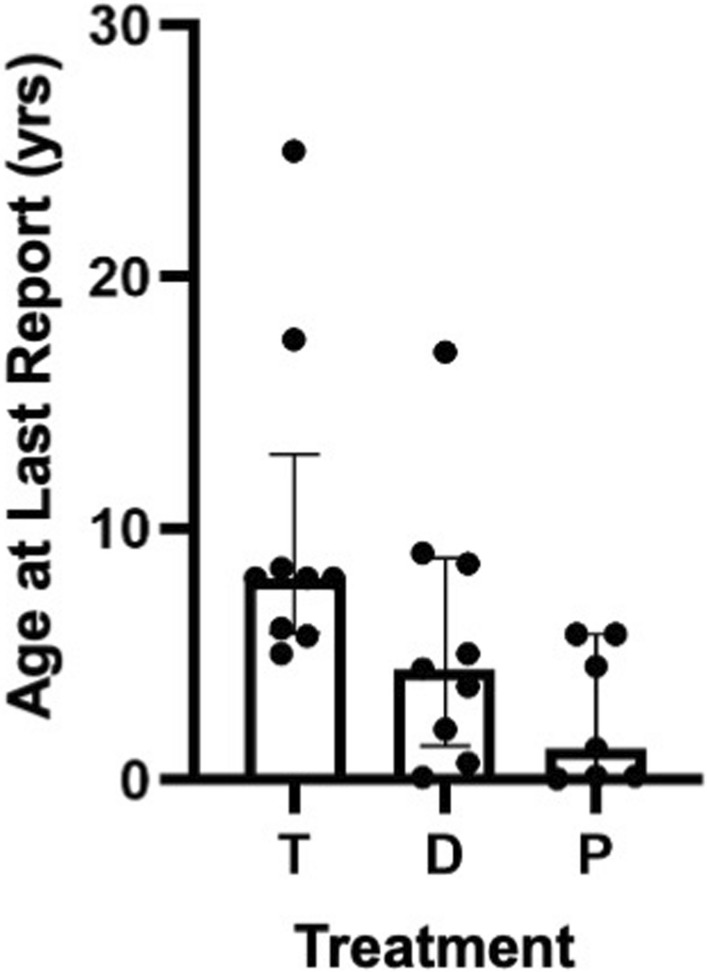
Overall survival at last report was 50%. Major influences on survival included: (1) age and organ involvement at first presentation; (2) receiving a kidney transplant, and (3) SGPL1 genotype. Among 48 SPLIS patients with nephropathy who had not received a kidney transplant, two clinical subgroups were distinguished. Of children diagnosed with SPLIS nephropathy before age one (n = 30), less than 30% were alive 2 years after diagnosis, and 17% were living at last report. Among those diagnosed at or after age one (n = 18), ~ 70% were alive 2 years after diagnosis, and 72% were living at time of last report. SPLIS patients homozygous for the SPL R222Q variant survived longer compared to patients with other genotypes. Kidney transplantation significantly extended survival outcomes.
Our results demonstrate that SPLIS is a phenotypically heterogeneous condition. We find that patients diagnosed with SPLIS nephropathy in the first year of life and patients presenting with prenatal findings represent two high-risk subgroups, whereas patients harboring the R222Q SGPL1 variant fare better than the rest. Time to progression from onset of proteinuria to end stage kidney disease varies from less than one month to five years, and kidney transplantation may be lifesaving.
神经鞘氨醇-1-磷酸裂解酶缺乏症(SPLIS)是一种新近认识到的代谢性遗传病,与类固醇耐药性肾病综合征以及肾上腺功能不全、免疫、神经和皮肤表现有关。SPLIS 是由编码依赖吡哆醛 5'磷酸的酶神经鞘氨醇-1-磷酸裂解酶的 SGPL1 基因中的失活突变引起的,该酶催化鞘脂代谢的最后一步。一些 SPLIS 患者接受了肾移植,另一些患者接受了维生素 B6 补充治疗。此外,包括基因治疗在内的靶向治疗正在临床前开发中。为了进行临床试验,对 SPLIS 的全貌和自然史进行特征描述将至关重要。我们对 76 例患者进行了回顾性分析,这些患者在首例患者中至少有一个提示性发现,并且通过分子遗传学检测确定了双等位基因 SGPL1 变体,从而确诊为 SPLIS。本研究的主要目的是确定影响 SPLIS 患者生存的因素。
截至最后报告时,总体生存率为 50%。对生存率有重大影响的因素包括:(1)首次就诊时的年龄和器官受累情况;(2)接受肾移植;(3)SGPL1 基因型。在 48 例未接受肾移植的 SPLIS 肾病患者中,区分出两个临床亚组。在 30 名在一岁之前被诊断为 SPLIS 肾病的儿童中,不到 30%的患者在诊断后 2 年仍然存活,17%的患者在最后一次报告时仍然存活。在那些在一岁或以上被诊断的患者中(n=18),大约 70%的患者在诊断后 2 年仍然存活,72%的患者在最后一次报告时仍然存活。与其他基因型的患者相比,纯合 SPL R222Q 变异的 SPLIS 患者的生存时间更长。肾移植显著延长了生存结果。
我们的研究结果表明,SPLIS 是一种表型异质性疾病。我们发现,在生命的第一年被诊断为 SPLIS 肾病的患者和有产前表现的患者代表了两个高风险亚组,而携带 SGPL1 R222Q 变异的患者比其他患者的预后更好。从蛋白尿发作到终末期肾病的进展时间从不到一个月到五年不等,肾移植可能是挽救生命的。