Centre for Inherited Diseases, Department of Research, Fondazione IRCCS Policlinico San Matteo, 27100 Pavia, Italy.
Neuroradiology Unit, Fondazione IRCCS Policlinico San Matteo, 27100 Pavia, Italy.
Genes (Basel). 2024 Sep 17;15(9):1212. doi: 10.3390/genes15091212.
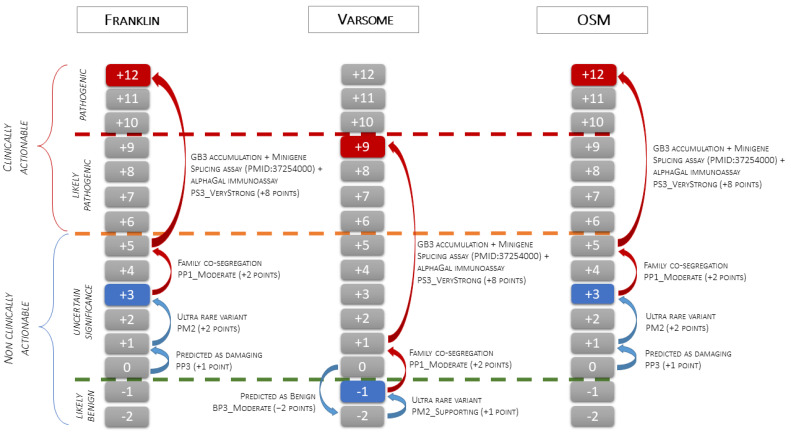
This study aims to demonstrate the role of case-level American College of Medical Genetics (ACMG) criteria, such as familial segregation and pathology data, in providing conclusive evidence for the pathogenicity of ultrarare variants causing Anderson-Fabry disease when gene-level and variant-level criteria provide ambiguous or discrepant results. Case/family description: A 52-year-old woman presented with new-onset shortness of breath, chest pain, and palpitations. Echocardiography revealed mild left ventricular wall thickening (14 mm) and mild diastolic dysfunction. She was the second of three siblings born to unrelated parents, both of whom died from malignancies. Family screening identified brothers, one affected 55-year-old with hypertension and asthma and one unaffected 47-year-old. The 15-year-old son of the proband complained of exercise-induced burning feet acral pain his electrocardiogram showed a short PR interval and signs of early hypertrophy.
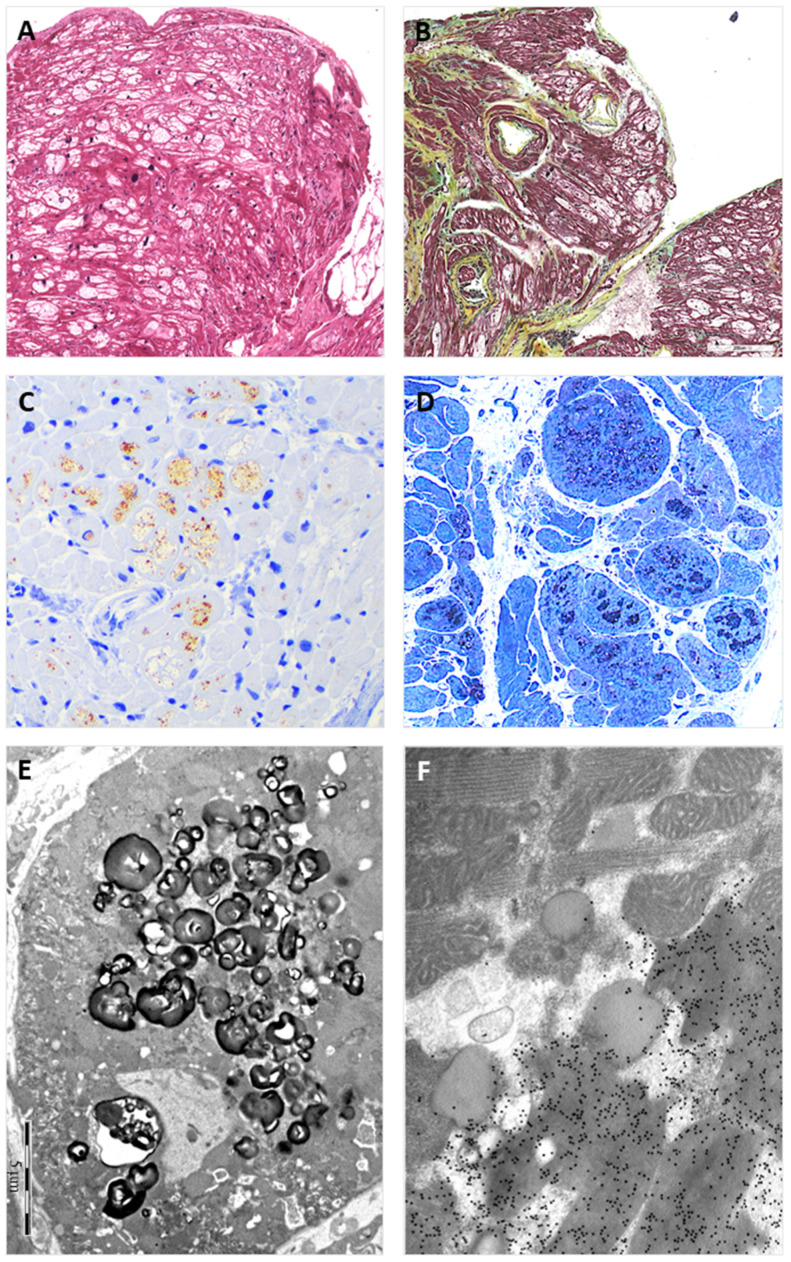
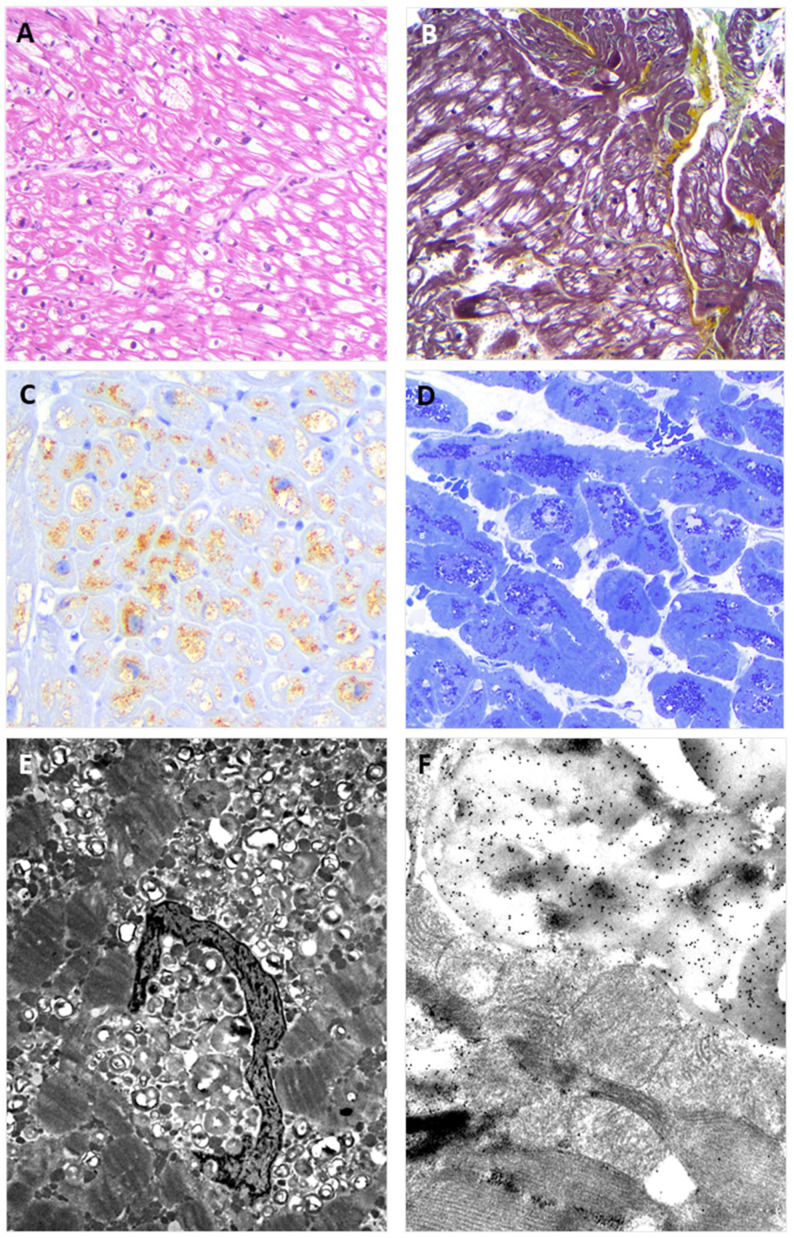
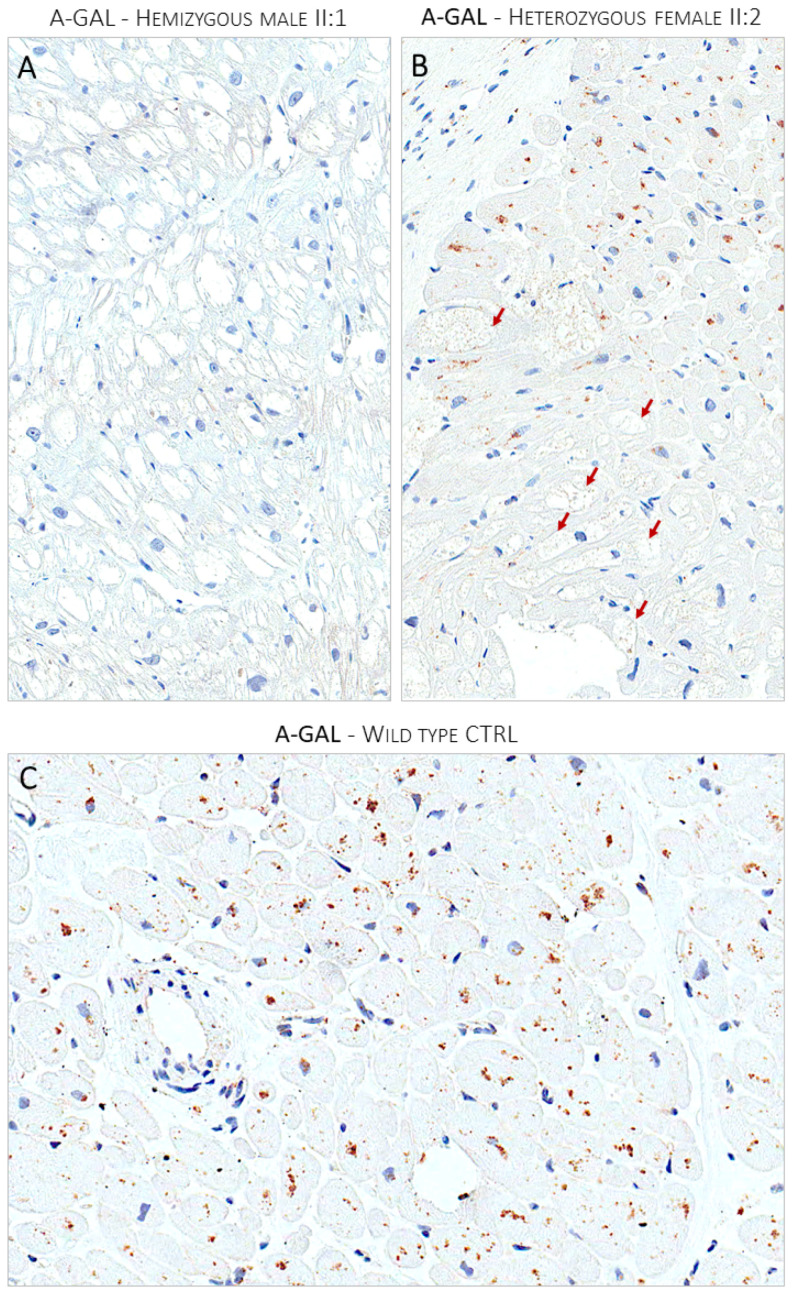
Endomyocardial biopsies of the proband and the affected sibling demonstrated substrate accumulation (globotriaosylceramide). The anti-α-galactosidase-A immunostain showed a total loss of the enzyme in the hemizygous male and a mosaic pattern in the heterozygous female. The next-generation sequencing short-read multigene panel identified the c.547+3A>G variant in the gene and excluded variants in other genes; Oxford-Nanopore long-read sequencing excluded known pathogenic deep intronic variants. A Multiplex-Ligation-dependent-Probe-Amplification assay excluded copy number variations. Based on the variant-level and gene-level ACMG criteria, the variant was classified as a Variant of Uncertain Significance or Likely Benign using different bioinformatic tools. By adding case-level functional data (endomyocardial biopsy, PS3_VeryStrong) and familial data (segregation of genotype with phenotype, PP2_Moderate), the variant was classified as Likely Pathogenic/Pathogenic.
ACMG case-level data can unambiguously resolve uncertain interpretations of variants.
本研究旨在展示病例级别的美国医学遗传学学院(ACMG)标准(如家族分离和病理学数据)在提供明确证据证明引起安德森-法布里病的超罕见变异的致病性方面的作用,当基因水平和变异水平的标准提供模棱两可或不一致的结果时。
病例/家族描述:一名 52 岁女性因新发呼吸急促、胸痛和心悸就诊。超声心动图显示左心室壁轻度增厚(14mm)和轻度舒张功能障碍。她是由无血缘关系的父母所生的三兄妹中的老二,父母均死于恶性肿瘤。家族筛查发现有两个兄弟,一个 55 岁的哥哥受高血压和哮喘影响,一个 47 岁的哥哥未受影响。先证者的 15 岁儿子抱怨运动引起的足部烧灼感,他的心电图显示 PR 间期缩短和早期肥厚的迹象。
先证者和受影响的兄弟的心肌活检显示底物堆积(半乳糖脑苷脂)。抗-α-半乳糖苷酶-A 免疫染色显示在半合子男性中酶完全缺失,在杂合子女性中呈现镶嵌模式。下一代测序短读长多基因面板在 基因中鉴定出 c.547+3A>G 变异,排除了其他基因中的变异;牛津-纳米孔长读测序排除了已知的致病性深内含子变异。多重连接依赖性探针扩增检测排除了拷贝数变异。基于变异水平和基因水平的 ACMG 标准,使用不同的生物信息学工具,该变异被归类为意义不明的变异或可能良性的变异。通过添加病例级别的功能数据(心肌活检、PS3_VeryStrong)和家族数据(基因型与表型的分离、PP2_Moderate),该变异被归类为可能致病性/致病性。
ACMG 病例级数据可以明确解决变异体不确定的解释。