Faculty of Medicine, Islamic University of Gaza, P.O. Box 108, Gaza, State of Palestine.
BMC Neurol. 2024 Sep 30;24(1):367. doi: 10.1186/s12883-024-03868-w.
Neuronal ceroid lipofuscinosis (NCL) is a heterogeneous group of 13 rare, progressive neurodegenerative diseases of the brain and retina. CLN14 is a very rare subtype of NCL caused by pathogenic variants in the KCTD7 gene. Only four cases of this subtype have been reported in the literature.
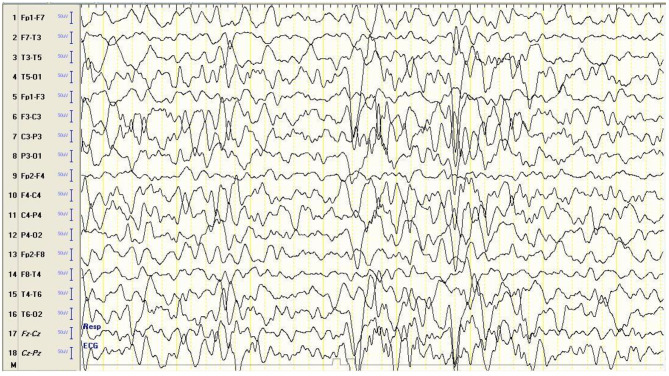
A nine-month-old, previously healthy male who was firstborn to first-cousin parents presented with progressive psychomotor regression, dysmorphic facial features, myoclonus, and vision loss. Neurological examination showed generalized hypotonia and brisk reflexes. He continued to deteriorate until age 18 months, when he developed his first generalized tonic-clonic seizure. An ophthalmological examination showed a hypopigmented fundus and slight temporal disc pallor. Brain MRI showed mild generalized brain atrophy and white matter disease. EEG revealed a severely abnormal trace marked by generalized, high amplitude, sharply contoured, polymorphic delta slowing intermixed with theta slowing and some alpha activity, with disorganized and scattered spikes and sharp waves. The patient continued to have uncontrolled seizures and further psychomotor regression until he died of status epilepticus and pneumonia at the age of 44 months. WES identified a novel homozygous variant c.413T > C, p.(Leu138Pro) in the KCTD7 gene, causing an amino acid transition from leucine to proline at position 138. Both parents were carriers of the same variant.
We present the fifth known case of CLN14 in the literature and report the clinical course and a novel underlying likely causative variant in the KCTD7 gene. The improving accessibility and affordability of genetic testing will likely uncover more NCL cases and further expand the disease's genotypic and phenotypic spectrum.
神经元蜡样脂褐质沉积症(NCL)是一组 13 种罕见的、进行性的脑和视网膜神经退行性疾病。CLN14 是由 KCTD7 基因突变引起的非常罕见的 NCL 亚型。该亚型在文献中仅报道过四例。
一名 9 月龄、此前健康的男婴,为表亲所生的第一胎,出现进行性精神运动倒退、畸形面容、肌阵挛和视力丧失。神经系统检查显示全身腱反射活跃。他持续恶化,直到 18 月龄时出现首次全面性强直阵挛发作。眼科检查显示眼底色素减退和轻微的视盘苍白。脑 MRI 显示轻度弥漫性脑萎缩和白质病变。EEG 显示严重异常的轨迹,表现为广泛性、高振幅、尖锐轮廓的多形性 δ 减速,伴有 θ 减速和一些 α 活动,伴有杂乱和分散的棘波和尖波。患儿持续出现无法控制的癫痫发作和进一步的精神运动倒退,最终因癫痫持续状态和肺炎于 44 月龄死亡。WES 鉴定出 KCTD7 基因中一个新的纯合变异 c.413T > C,p.(Leu138Pro),导致第 138 位的亮氨酸突变为脯氨酸。父母均为该变异的携带者。
我们报告了文献中第五例已知的 CLN14 病例,并报道了临床病程和 KCTD7 基因中一个新的潜在致病变异。遗传检测的可及性和可负担性的提高可能会发现更多的 NCL 病例,并进一步扩大该疾病的基因型和表型谱。