Interdisciplinary Program in Bioinformatics, Seoul National University, Seoul, 08826, Republic of Korea.
AIGENDRUG Co., Ltd, Seoul, Republic of Korea.
Sci Rep. 2024 Oct 3;14(1):23010. doi: 10.1038/s41598-024-72868-0.
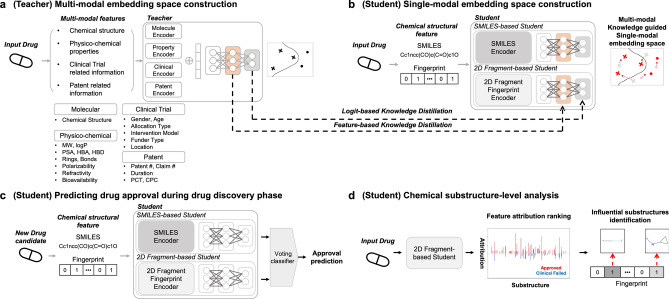
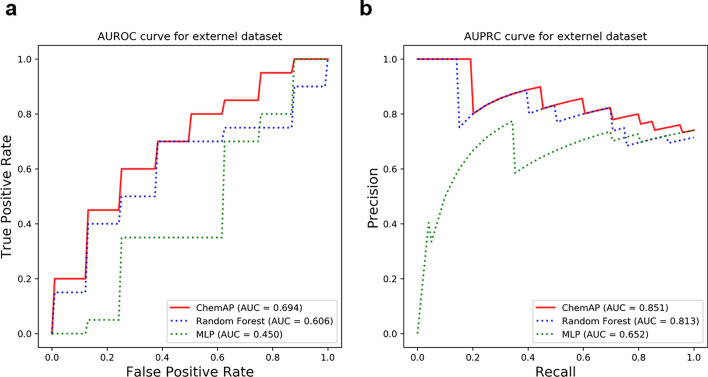
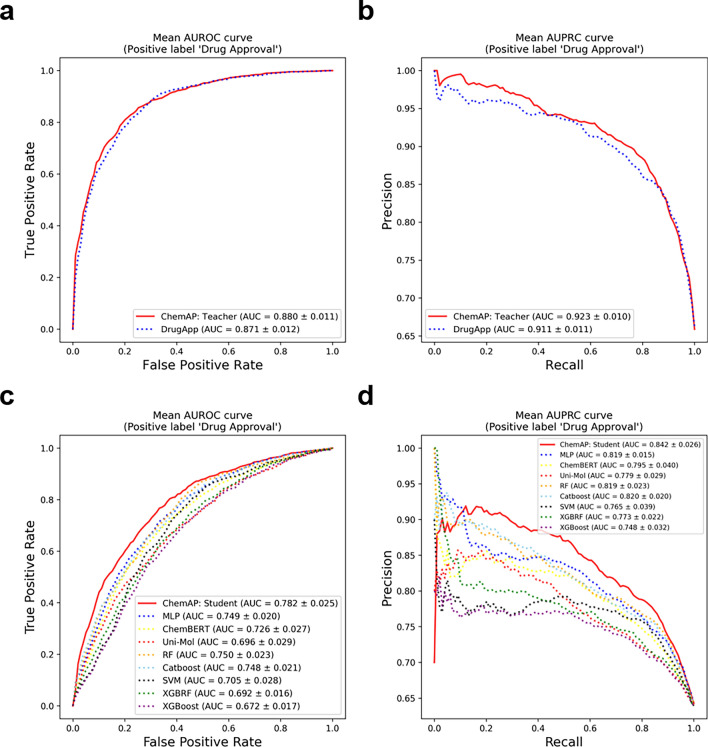
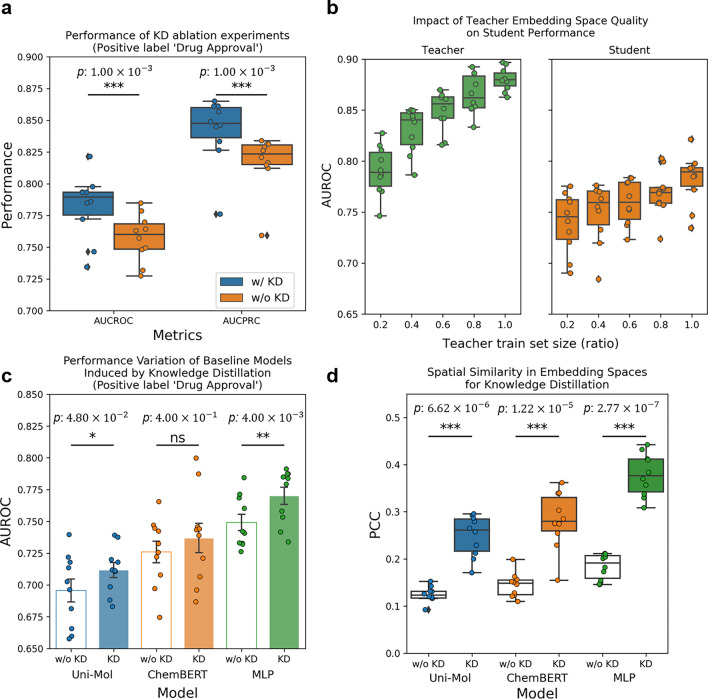
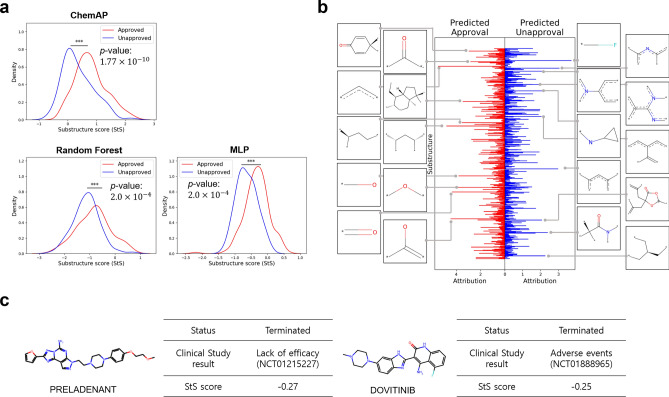
Recent studies showed that the likelihood of drug approval can be predicted with clinical data and structure information of drug using computational approaches. Predicting the likelihood of drug approval can be innovative and of high impact. However, models that leverage clinical data are applicable only in clinical stages, which is not very practical. Prioritizing drug candidates and early-stage decision-making in the de novo drug development process is crucial in pharmaceutical research to optimize resource allocation. For early-stage decision-making, we need a computational model that uses only chemical structures. This seemingly impossible task may utilize the predictive power with multi-modal features including clinical data. In this work, we introduce ChemAP (Chemical structure-based drug Approval Predictor), a novel deep learning scheme for drug approval prediction in the early-stage drug discovery phase. ChemAP aims to enhance the possibility of early-stage decision-making by enriching semantic knowledge to fill in the gap between multi-modal and single-modal chemical spaces through knowledge distillation techniques. This approach facilitates the effective construction of chemical space solely from chemical structure data, guided by multi-modal knowledge related to efficacy, such as clinical trials and patents of drugs. In this study, ChemAP achieved state-of-the-art performance, outperforming both traditional machine learning and deep learning models in drug approval prediction, with AUROC and AUPRC scores of 0.782 and 0.842 respectively on the drug approval benchmark dataset. Additionally, we demonstrated its generalizability by outperforming baseline models on a recent external dataset, which included drugs from the 2023 FDA-approved list and the 2024 clinical trial failure drug list, achieving AUROC and AUPRC scores of 0.694 and 0.851. These results demonstrate that ChemAP is an effective method in predicting drug approval only with chemical structure information of drug so that decision-making can be done at the early stages of drug development process. To the best of our knowledge, our work is the first of its kind to show that prediction of drug approval is possible only with structure information of drug by defining the chemical space of approved and unapproved drugs using deep learning technology.
最近的研究表明,使用计算方法可以通过药物的临床数据和结构信息来预测药物的批准可能性。预测药物的批准可能性具有创新性和高影响力。然而,利用临床数据的模型仅适用于临床阶段,这并不十分实用。在新药开发过程中,对药物候选物进行优先级排序和早期决策对于药物研究至关重要,以优化资源分配。对于早期决策,我们需要一个仅使用化学结构的计算模型。这项看似不可能的任务可能会利用包括临床数据在内的多模态特征的预测能力。在这项工作中,我们引入了 ChemAP(基于化学结构的药物批准预测器),这是一种用于早期药物发现阶段药物批准预测的新型深度学习方案。ChemAP 旨在通过知识蒸馏技术,丰富语义知识,在多模态和单模态化学空间之间架起桥梁,从而增强早期决策的可能性。这种方法通过利用与疗效相关的多模态知识(如临床试验和药物专利),从化学结构数据中有效地构建化学空间。在这项研究中,ChemAP 在药物批准基准数据集上实现了最先进的性能,在药物批准预测方面优于传统机器学习和深度学习模型,AUROC 和 AUPRC 得分分别为 0.782 和 0.842。此外,我们通过在最近的外部数据集上优于基线模型来证明其泛化能力,该数据集包括 2023 年 FDA 批准的药物清单和 2024 年临床试验失败药物清单中的药物,AUROC 和 AUPRC 得分分别为 0.694 和 0.851。这些结果表明,ChemAP 是一种仅使用药物的化学结构信息预测药物批准的有效方法,从而可以在药物开发过程的早期阶段做出决策。据我们所知,我们的工作是第一个表明仅通过使用深度学习技术定义已批准和未批准药物的化学空间,仅使用药物的结构信息就可以预测药物批准的工作。