Laboratory for Cardiovascular Genomics and Informatics, RIKEN Center for Integrative Medical Sciences, Yokohama, Japan.
Medical and Population Genetics and Cardiovascular Disease Initiative, Broad Institute of MIT and Harvard, Cambridge, MA, USA.
Nat Genet. 2024 Oct;56(10):2027-2035. doi: 10.1038/s41588-024-01913-5. Epub 2024 Oct 3.
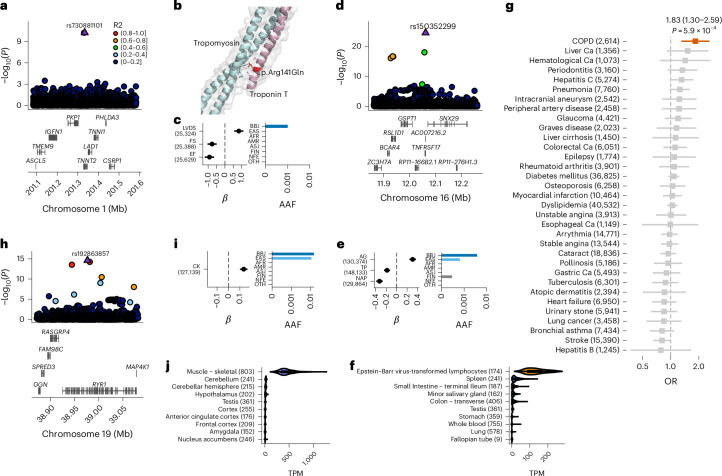
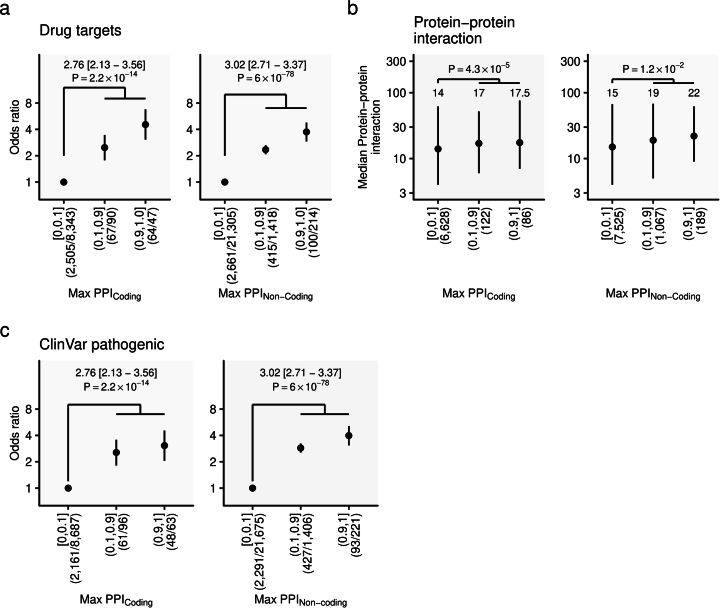
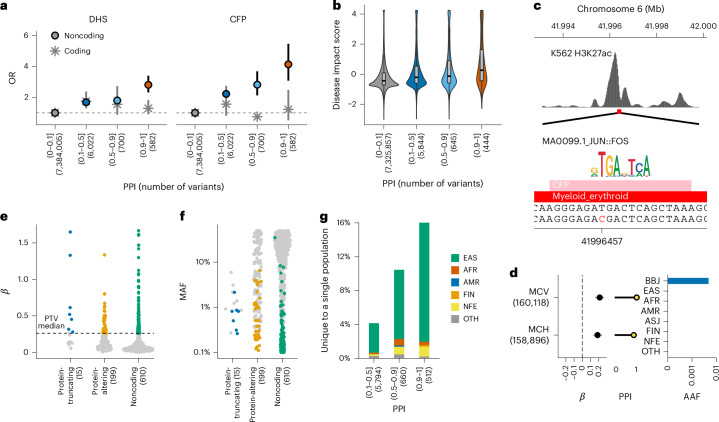
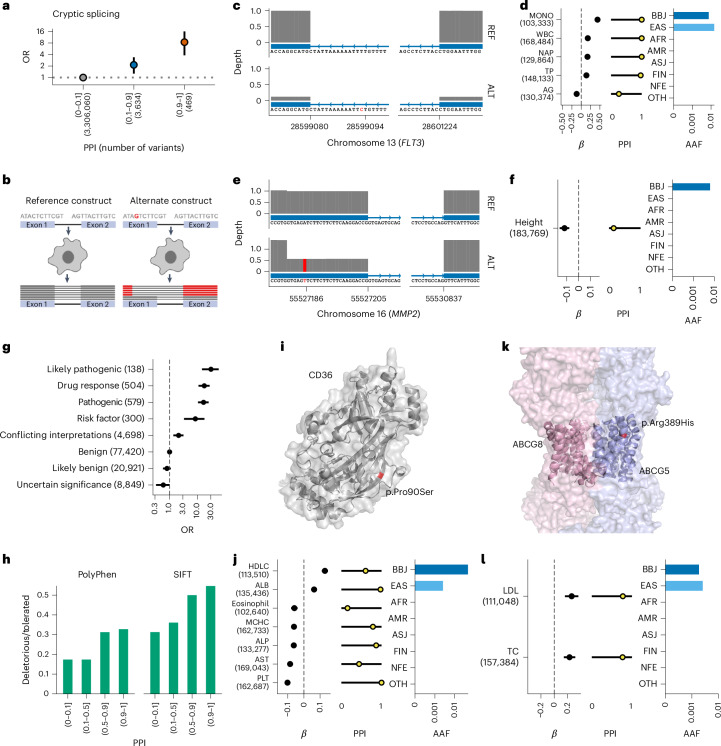
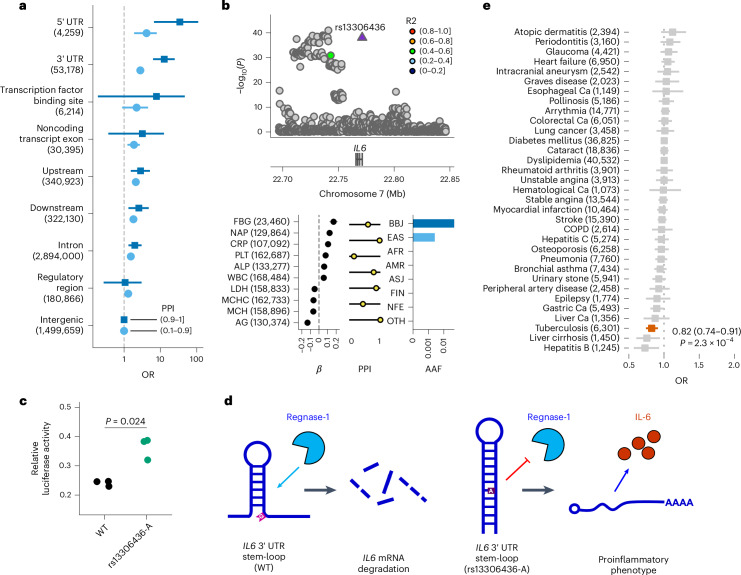
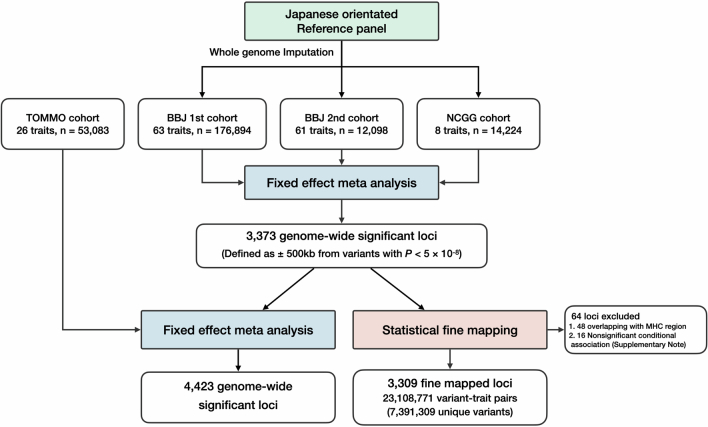
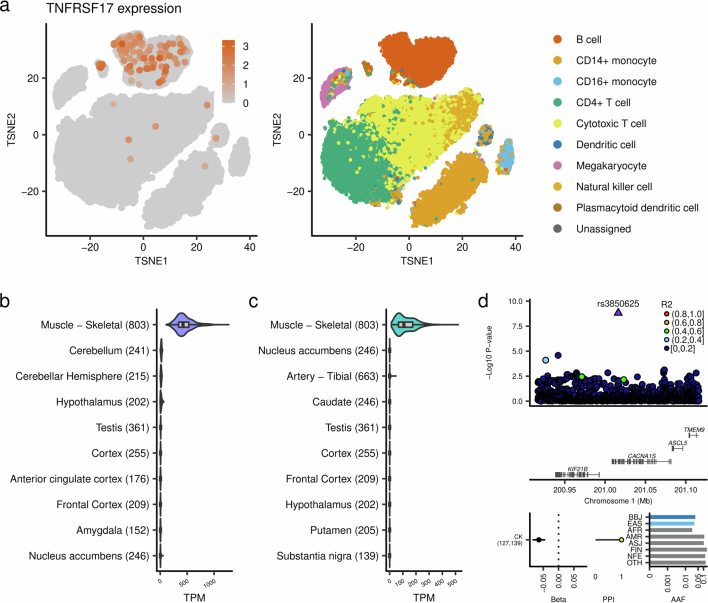
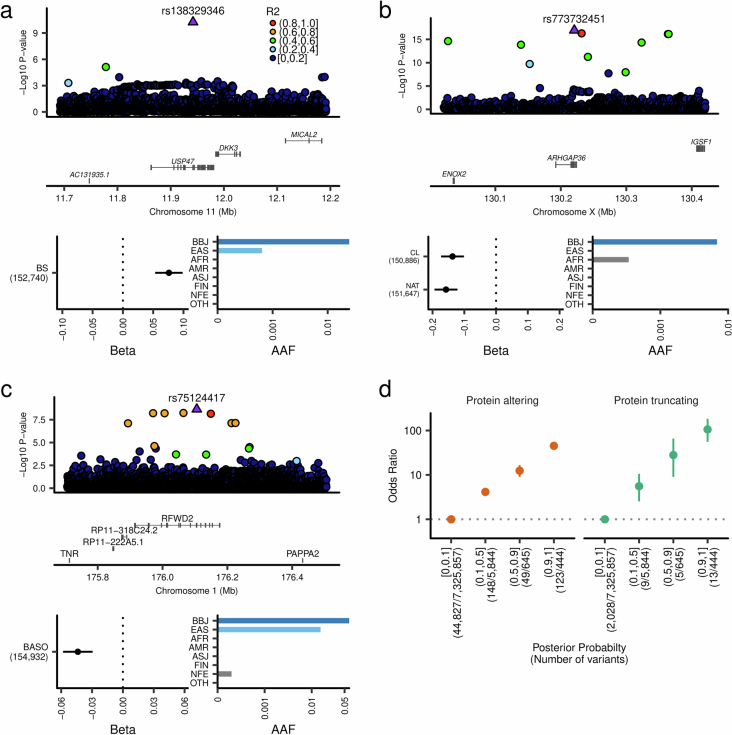
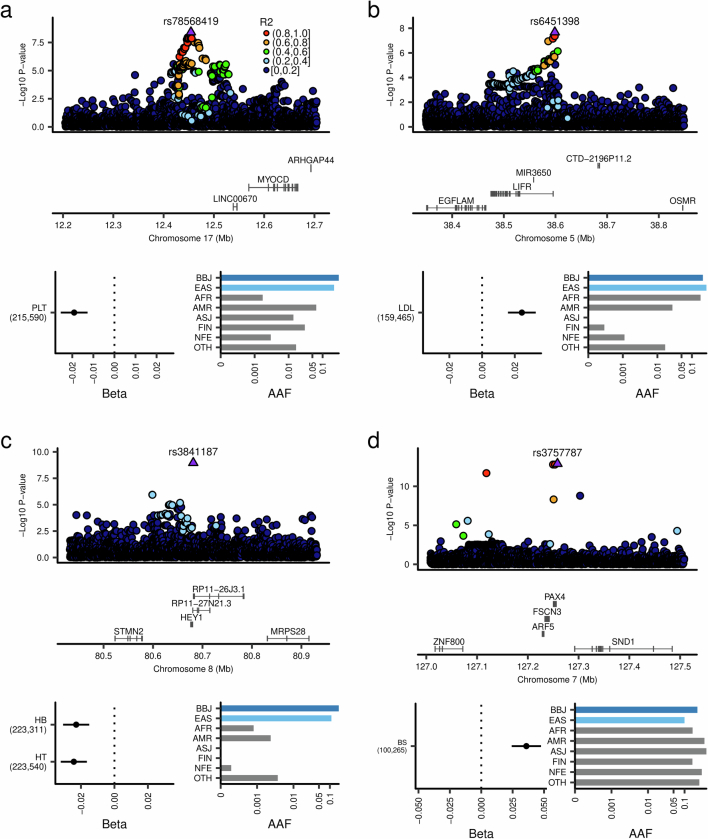
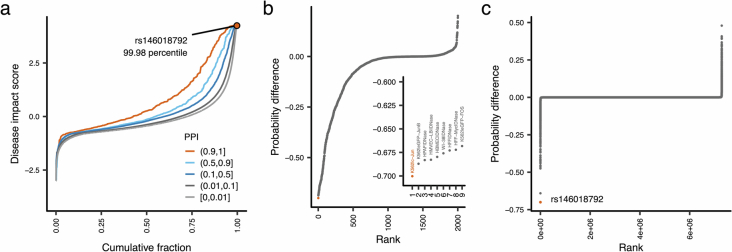
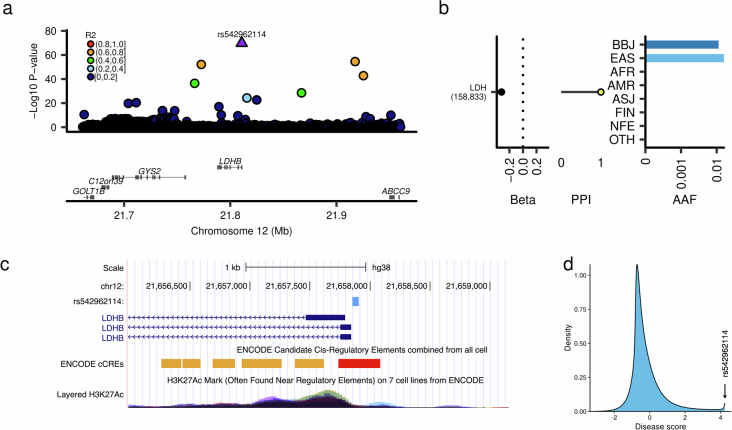
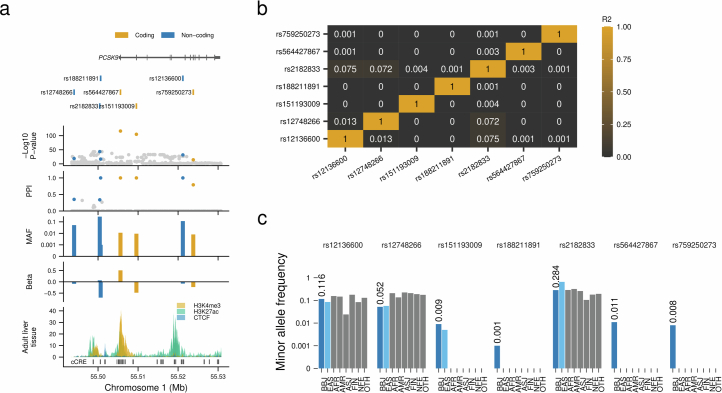
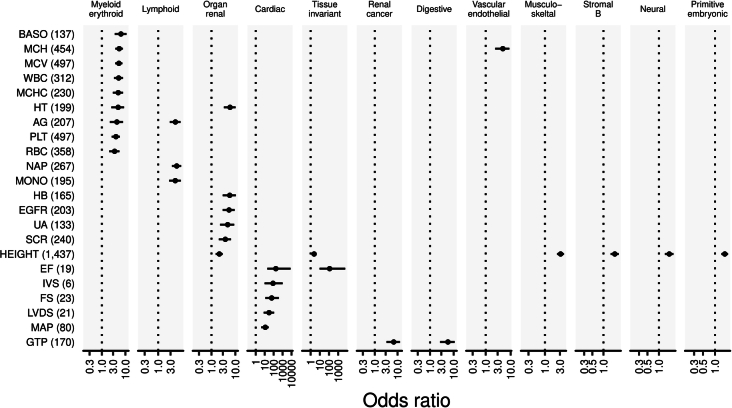
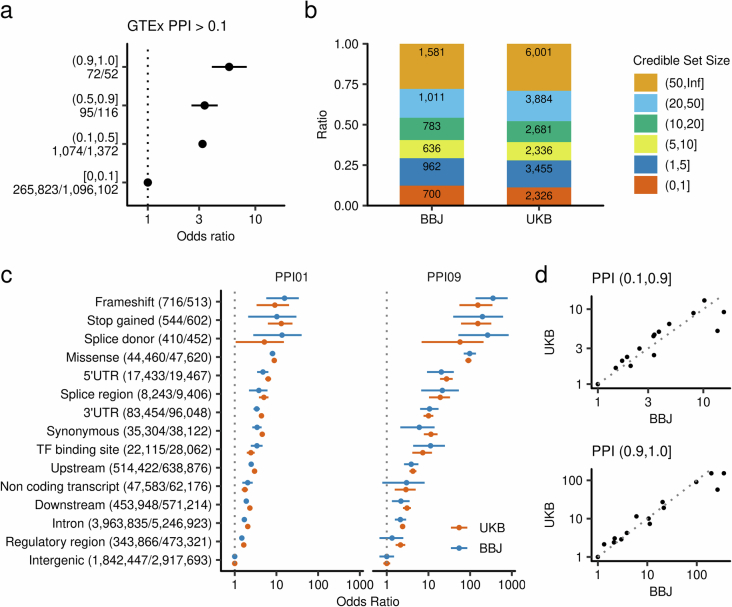
Human genetic variants are associated with many traits through largely unknown mechanisms. Here, combining approximately 260,000 Japanese study participants, a Japanese-specific genotype reference panel and statistical fine-mapping, we identified 4,423 significant loci across 63 quantitative traits, among which 601 were new, and 9,406 putatively causal variants. New associations included Japanese-specific coding, splicing and noncoding variants, exemplified by a damaging missense variant rs730881101 in TNNT2 associated with lower heart function and increased risk for heart failure (P = 1.4 × 10 and odds ratio = 4.5, 95% confidence interval = 3.1-6.5). Putative causal noncoding variants were supported by state-of-art in silico functional assays and had comparable effect sizes to coding variants. A plausible example of new mechanisms of causal variants is an enrichment of causal variants in 3' untranslated regions (UTRs), including the Japanese-specific rs13306436 in IL6 associated with pro-inflammatory traits and protection against tuberculosis. We experimentally showed that transcripts with rs13306436 are resistant to mRNA degradation by regnase-1, an RNA-binding protein. Our study provides a list of fine-mapped causal variants to be tested for functionality and underscores the importance of sequencing, genotyping and association efforts in diverse populations.
人类遗传变异通过很大程度上未知的机制与许多特征相关。在这里,我们结合了大约 260,000 名日本研究参与者、一个日本特有的基因型参考面板和统计精细映射,在 63 个定量特征中确定了 4,423 个显著位点,其中 601 个是新的,9,406 个是推定的因果变异。新的关联包括日本特有的编码、剪接和非编码变异,例如在 TNNT2 中的破坏性错义变异 rs730881101 与较低的心脏功能和心力衰竭风险增加相关(P=1.4×10 和优势比=4.5,95%置信区间=3.1-6.5)。推定的因果非编码变异得到了最先进的计算机功能测定的支持,并且与编码变异具有可比的效应大小。因果变异新机制的一个合理例子是在 3'非翻译区(UTR)中因果变异的富集,包括与促炎特征和结核保护相关的日本特有的 rs13306436 在 IL6 中的富集。我们通过实验表明,携带 rs13306436 的转录本不易被 RNA 结合蛋白 regnase-1 降解。我们的研究提供了一份精细映射的因果变异列表,以供测试其功能,并强调了在不同人群中进行测序、基因分型和关联研究的重要性。