MOE Medical Basic Research Innovation Center for Gut Microbiota and Chronic Diseases, School of Medicine, Jiangnan University, Wuxi, 214122, China.
Department of Physiology, Eberhard-Karls-University of Tübingen, Tübingen University, Tübingen, 72076, Germany.
Cell Commun Signal. 2024 Oct 11;22(1):488. doi: 10.1186/s12964-024-01873-7.
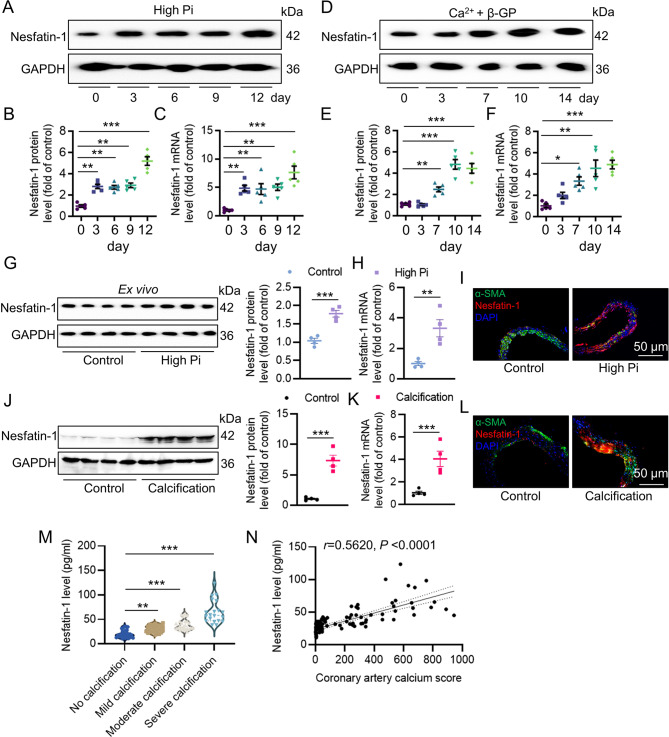
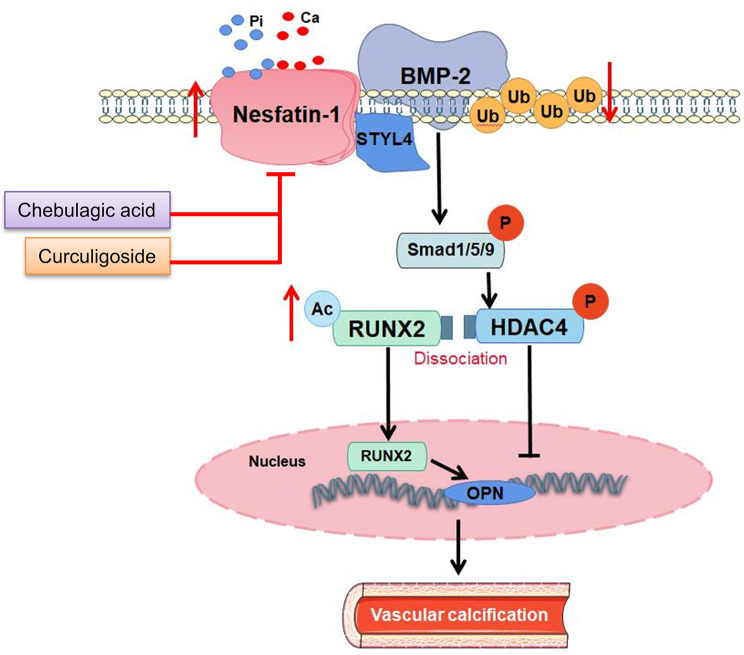
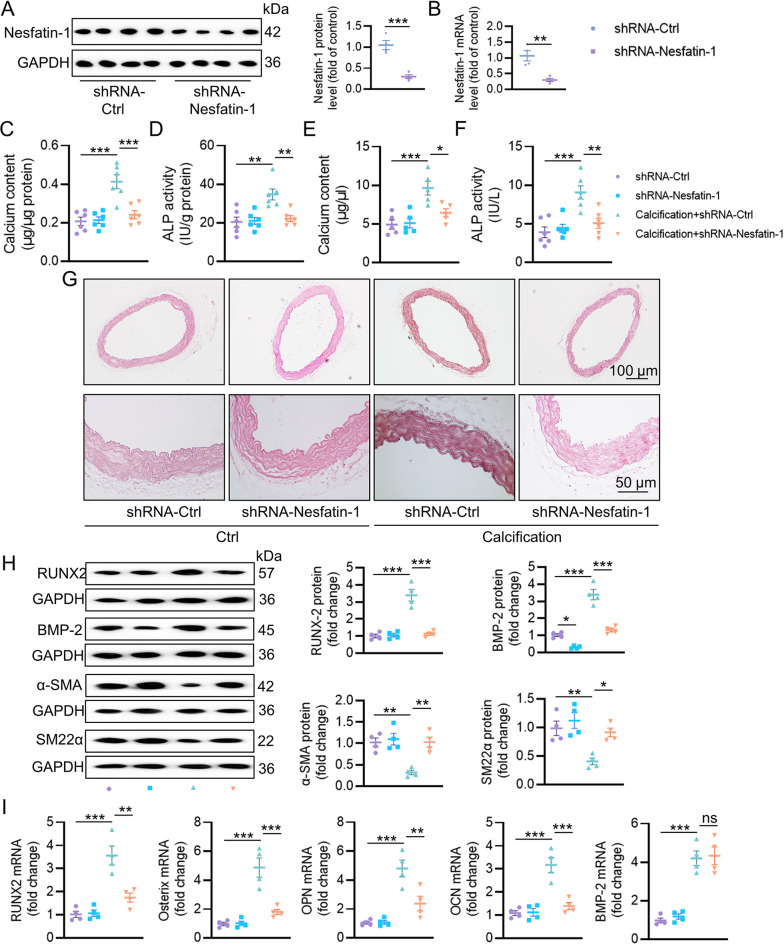
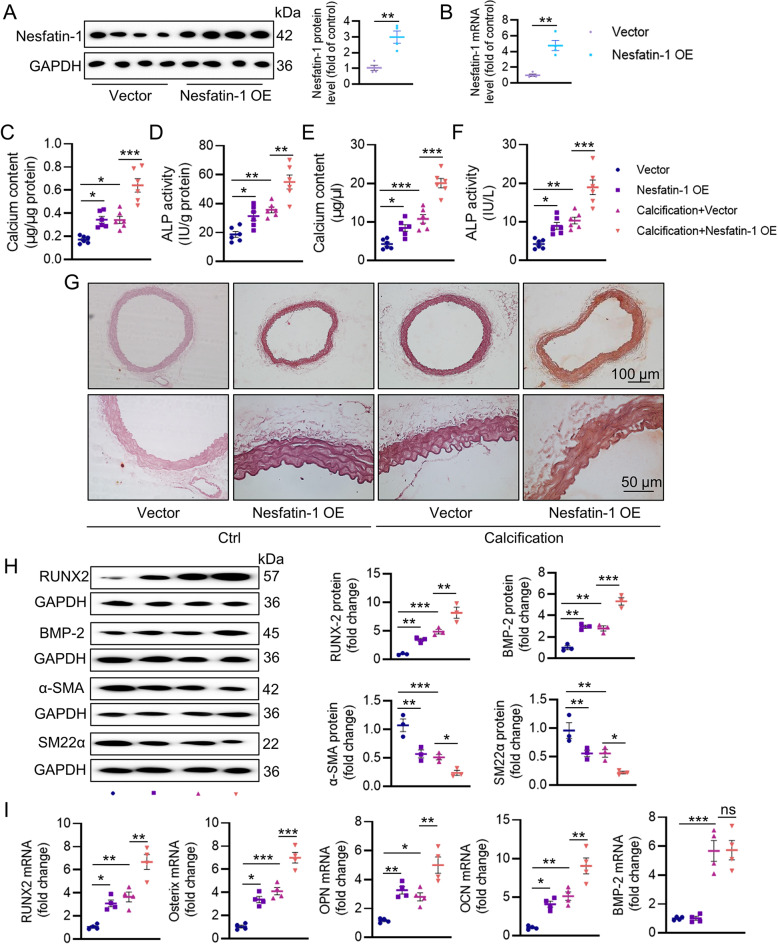
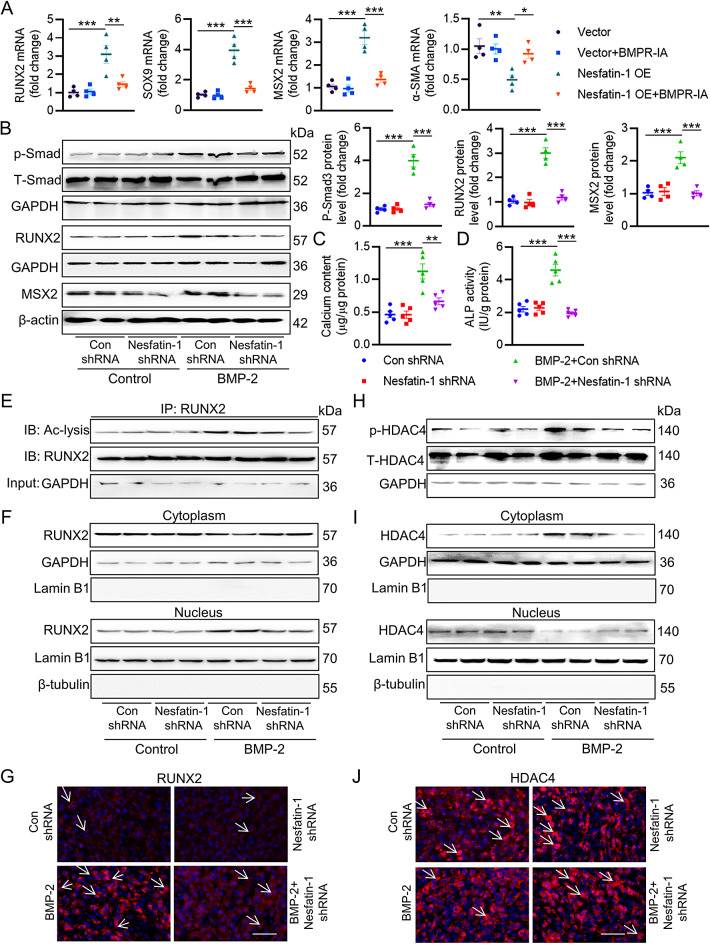
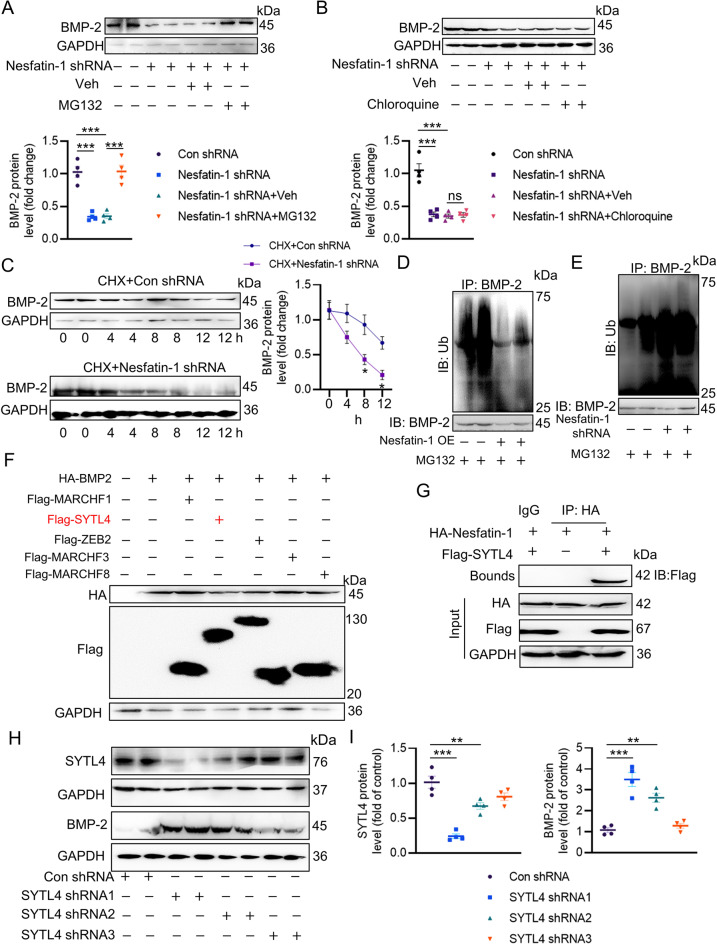
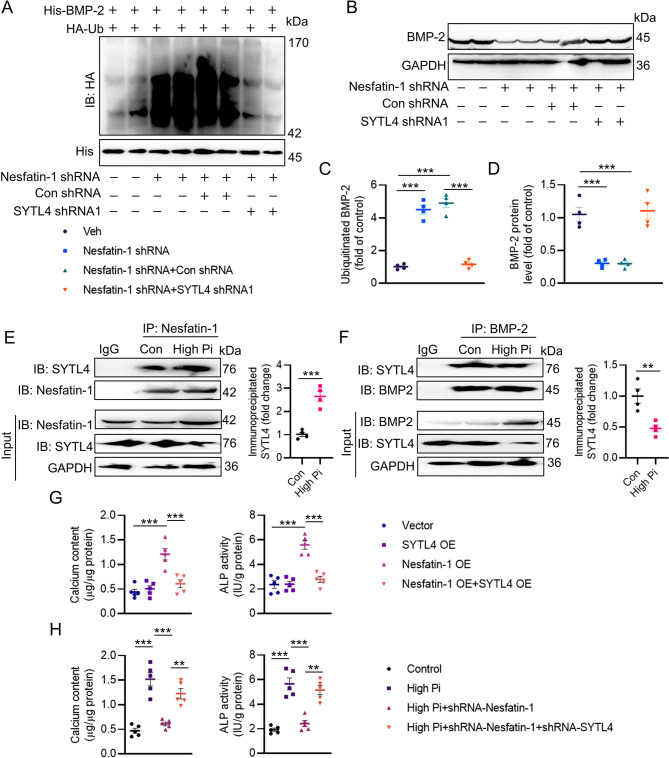
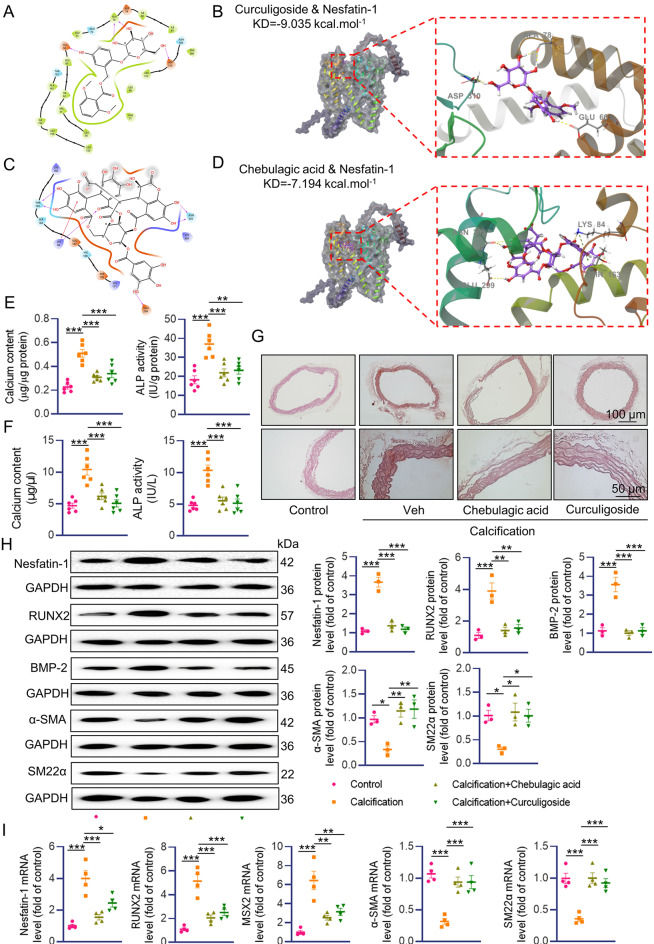
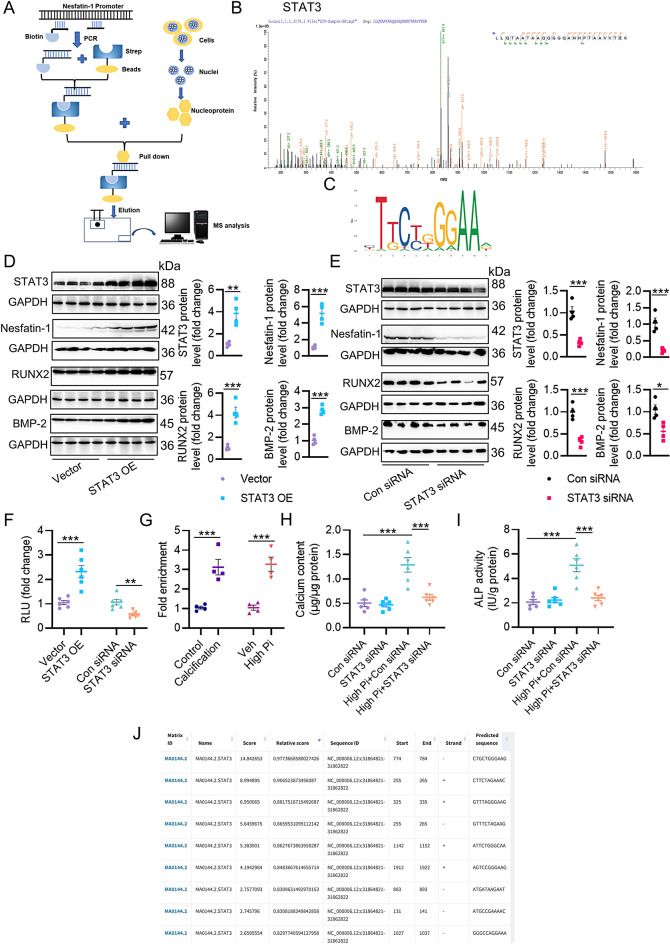
Vascular calcification (VC) arises from the accumulation of calcium salts in the intimal or tunica media layer of the aorta, contributing to higher risk of cardiovascular events and mortality. Despite this, the mechanisms driving VC remain incompletely understood. We previously described that nesfatin-1 functioned as a switch for vascular smooth muscle cells (VSMCs) plasticity in hypertension and neointimal hyperplasia. In this study, we sought to investigate the role and mechanism of nesfatin-1 in VC. The expression of nesfatin-1 was measured in calcified VSMCs and aortas, as well as in patients. Loss- and gain-of-function experiments were evaluated the roles of nesfatin-1 in VC pathogenesis. The transcription activation of nesfatin-1 was detected using a mass spectrometry. We found higher levels of nesfatin-1 in both calcified VSMCs and aortas, as well as in patients with coronary calcification. Loss-of-function and gain-of-function experiments revealed that nesfatin-1 was a key regulator of VC by facilitating the osteogenic transformation of VSMCs. Mechanistically, nesfatin-1 promoted the de-ubiquitination and stability of BMP-2 via inhibiting the E3 ligase SYTL4, and the interaction of nesfatin-1 with BMP-2 potentiated BMP-2 signaling and induced phosphorylation of Smad, followed by HDAC4 phosphorylation and nuclear exclusion. The dissociation of HDAC4 from RUNX2 elicited RUNX2 acetylation and subsequent nuclear translocation, leading to the transcription upregulation of OPN, a critical player in VC. From a small library of natural compounds, we identified that Curculigoside and Chebulagic acid reduced VC development via binding to and inhibiting nesfatin-1. Eventually, we designed a mass spectrometry-based DNA-protein interaction screening to identify that STAT3 mediated the transcription activation of nesfatin-1 in the context of VC. Overall, our study demonstrates that nesfatin-1 enhances BMP-2 signaling by inhibiting the E3 ligase SYTL4, thereby stabilizing BMP-2 and facilitating the downstream phosphorylation of SMAD1/5/9 and HDAC4. This signaling cascade leads to RUNX2 activation and the transcriptional upregulation of MSX2, driving VC. These insights position nesfatin-1 as a potential therapeutic target for preventing or treating VC, advancing our understanding of the molecular mechanisms underlying this critical cardiovascular condition.
血管钙化(VC)是指钙盐在主动脉内膜或中膜层的积累,增加了心血管事件和死亡率的风险。尽管如此,驱动 VC 的机制仍不完全清楚。我们之前描述过, nesfatin-1 在高血压和新生内膜增生中作为血管平滑肌细胞(VSMCs)可塑性的开关。在这项研究中,我们旨在研究 nesfatin-1 在 VC 中的作用和机制。测量了钙化 VSMCs 和主动脉以及患者中 nesfatin-1 的表达。通过失活和激活实验评估了 nesfatin-1 在 VC 发病机制中的作用。通过质谱法检测 nesfatin-1 的转录激活。我们发现,在钙化的 VSMCs 和主动脉以及患有冠状动脉钙化的患者中, nesfatin-1 的水平更高。失活和激活实验表明, nesfatin-1 通过促进 VSMCs 的成骨转化,是 VC 的关键调节因子。从机制上讲, nesfatin-1 通过抑制 E3 连接酶 SYTL4 促进 BMP-2 的去泛素化和稳定性, nesfatin-1 与 BMP-2 的相互作用增强了 BMP-2 信号,并诱导 Smad 的磷酸化,随后 HDAC4 的磷酸化和核排斥。HDAC4 从 RUNX2 上的解离引起 RUNX2 的乙酰化和随后的核转位,导致 VC 中的关键参与者 OPN 的转录上调。从小的天然化合物文库中,我们发现, Curculigoside 和 Chebulagic acid 通过与 nesfatin-1 结合并抑制其活性,减少了 VC 的发展。最终,我们设计了基于质谱的 DNA-蛋白相互作用筛选,以确定 STAT3 在 VC 背景下介导 nesfatin-1 的转录激活。总的来说,我们的研究表明, nesfatin-1 通过抑制 E3 连接酶 SYTL4 增强 BMP-2 信号,从而稳定 BMP-2 并促进 SMAD1/5/9 和 HDAC4 的下游磷酸化。这个信号级联导致 RUNX2 的激活和 MSX2 的转录上调,从而驱动 VC。这些发现将 nesfatin-1 定位为预防或治疗 VC 的潜在治疗靶点,推进了我们对这种关键心血管疾病的分子机制的理解。