Department of Infectious Diseases, Institute of Biomedicine, The Sahlgrenska Academy, University of Gothenburg, Guldhedsgatan 10A, Gothenburg, 413 46, Sweden.
Division of Systems and Synthetic Biology, Department of Life Sciences, SciLifeLab, Chalmers University of Technology, Gothenburg, 412 96, Sweden.
BMC Genomics. 2024 Oct 14;25(1):959. doi: 10.1186/s12864-024-10876-0.
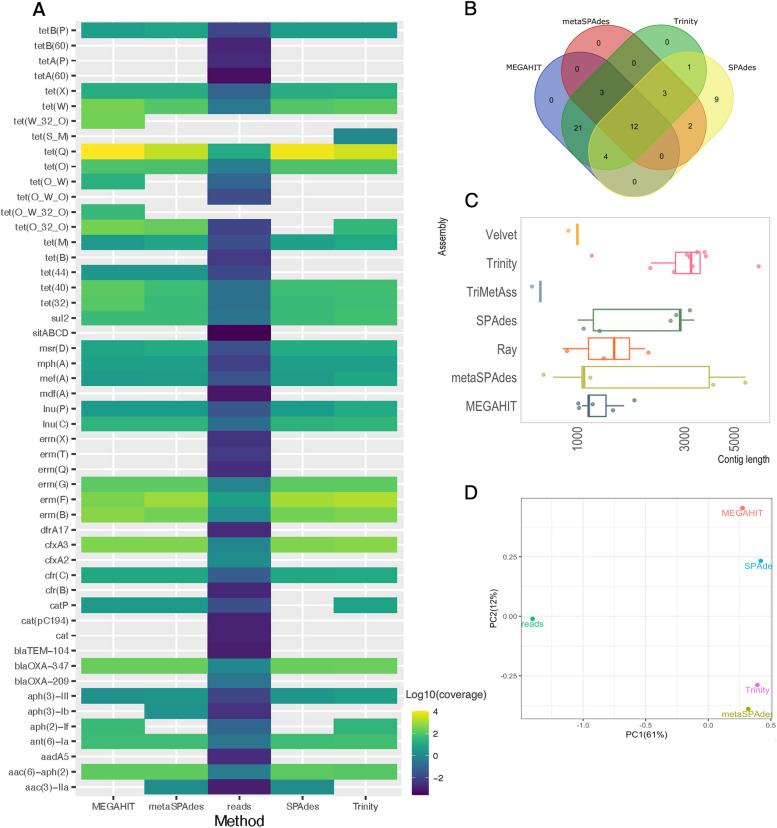
Assembly of metagenomic samples can provide essential information about the mobility potential and taxonomic origin of antibiotic resistance genes (ARGs) and inform interventions to prevent further spread of resistant bacteria. However, similar to other conserved regions, such as ribosomal RNA genes and mobile genetic elements, almost identical ARGs typically occur in multiple genomic contexts across different species, representing a considerable challenge for the assembly process. Usually, this results in many fragmented contigs of unclear origin, complicating the risk assessment of ARG detections. To systematically investigate the impact of this issue on detection, quantification and contextualization of ARGs, we evaluated the performance of different assembly approaches, including genomic-, metagenomic- and transcriptomic-specialized assemblers. We quantified recovery and accuracy rates of each tool for ARGs both from in silico spiked metagenomic samples as well as real samples sequenced using both long- and short-read sequencing technologies.
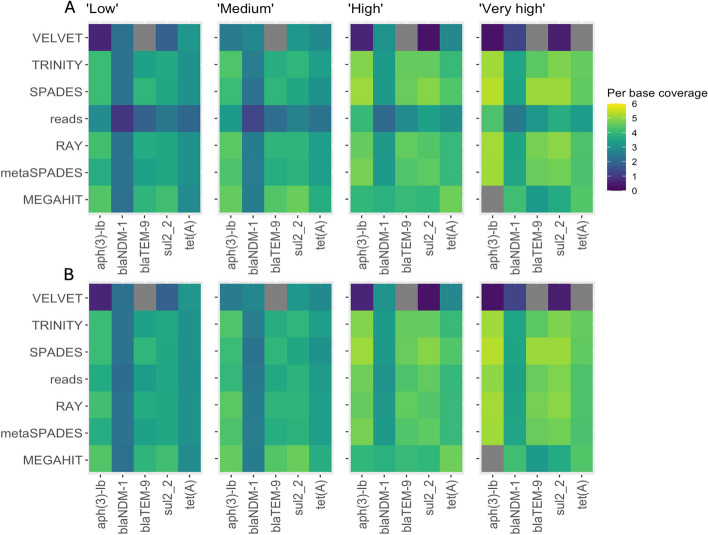
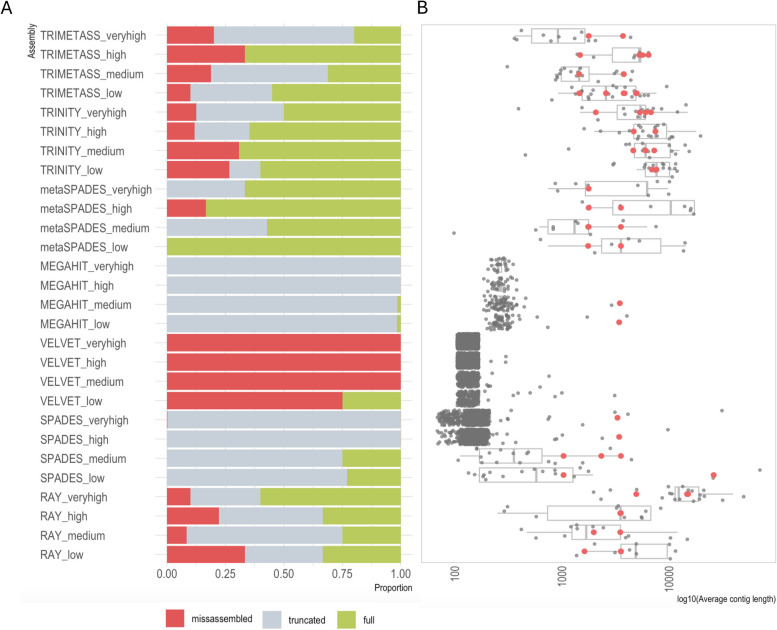
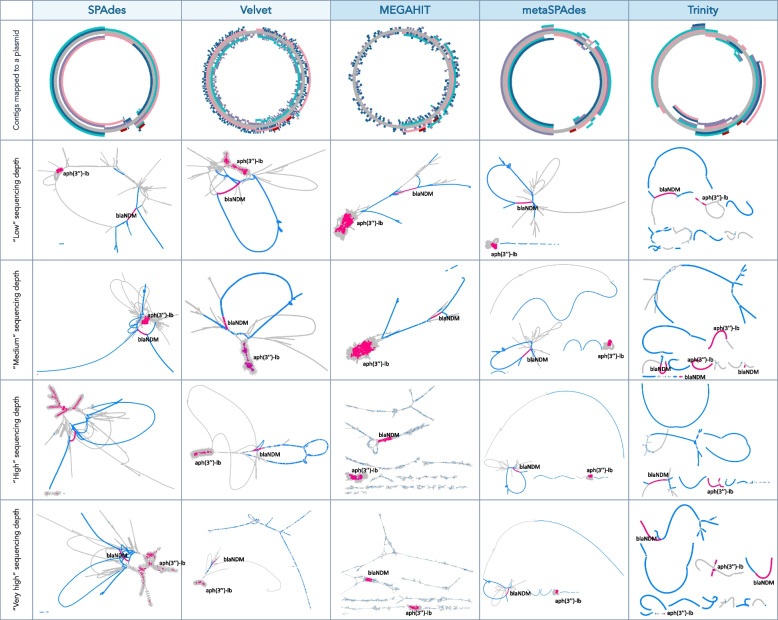
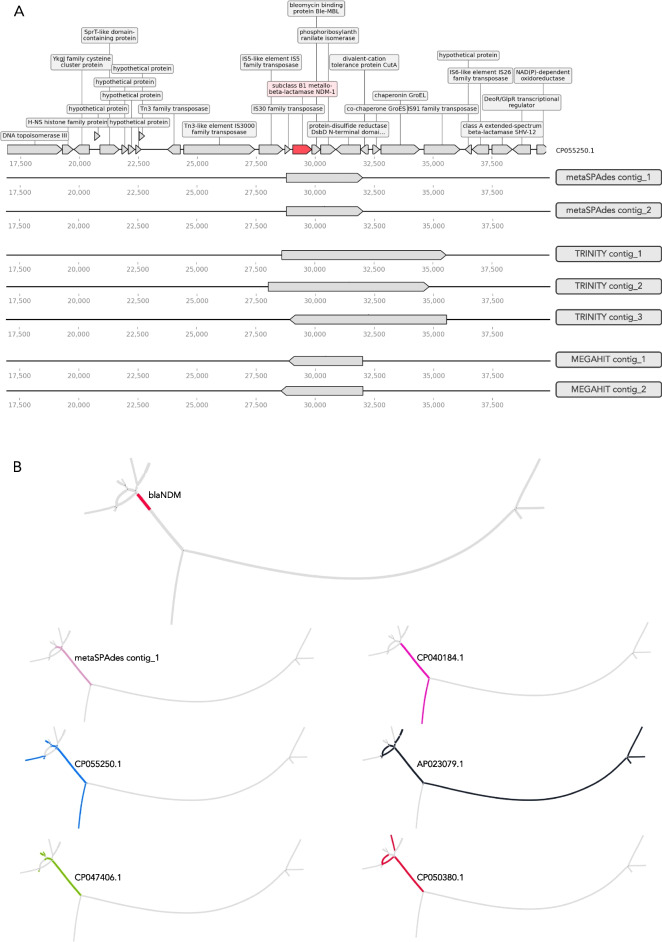
The results revealed that none of the investigated tools can accurately capture genomic contexts present in samples of high complexity. The transcriptomic assembler Trinity showed a better performance in terms of reconstructing longer and fewer contigs matching unique genomic contexts, which can be beneficial for deciphering the taxonomic origin of ARGs. The currently commonly used metagenomic assembly tools metaSPAdes and MEGAHIT were able to identify the ARG repertoire but failed to fully recover the diversity of genomic contexts present in a sample. On top of that, in a complex scenario MEGAHIT produced very short contigs, which can lead to considerable underestimation of the resistome in a given sample.
Our study shows that metaSPAdes and Trinity would be the preferable tools in terms of accuracy to recover correct genomic contexts around ARGs in metagenomic samples characterized by uneven coverages. Overall, the inability of assemblers to reconstruct long ARG-containing contigs has impacts on ARG quantification, suggesting that directly mapping reads to an ARG database should be performed as a complementary strategy to get accurate ARG abundance and diversity measures.
组装宏基因组样本可以提供有关抗生素抗性基因(ARGs)的移动潜力和分类起源的重要信息,并为防止耐药细菌进一步传播提供干预措施。然而,与核糖体 RNA 基因和移动遗传元件等其他保守区域类似,几乎相同的 ARGs 通常存在于不同物种的多个基因组环境中,这对组装过程构成了相当大的挑战。通常,这会导致许多来源不明的碎片化 contigs,从而使 ARG 检测的风险评估变得复杂。为了系统地研究这个问题对 ARG 检测、定量和语境化的影响,我们评估了不同组装方法的性能,包括基因组、宏基因组和转录组专业化组装器。我们量化了每个工具对来自模拟宏基因组样本和使用长读和短读测序技术测序的真实样本中 ARGs 的恢复率和准确性。
结果表明,没有一种研究工具可以准确捕获高复杂性样本中的基因组环境。转录组组装器 Trinity 在重建与独特基因组环境匹配的更长和更少 contigs 方面表现出更好的性能,这有助于解析 ARG 的分类起源。目前常用的宏基因组组装工具 metaSPAdes 和 MEGAHIT 能够识别 ARG 库,但无法完全恢复样本中存在的基因组环境多样性。除此之外,在复杂情况下,MEGAHIT 产生的 contigs 非常短,这可能导致给定样本中的抗药性基因被严重低估。
我们的研究表明,在覆盖不均匀的宏基因组样本中,metaSPAdes 和 Trinity 在准确性方面更适合恢复正确的 ARG 周围的基因组环境。总体而言,组装器无法构建长的含 ARG 的 contigs 会对 ARG 定量产生影响,这表明应该将读取直接映射到 ARG 数据库,作为获得准确的 ARG 丰度和多样性测量的补充策略。