Liu Yuemin, Gao Yunxiang, Altalhi Tariq, Liu Di-Jia, Yakobson Boris I
Department of Chemistry and Physics, Prairie View A&M University, Prairie View, TX 77446, USA.
Department of Chemistry, Rice University, Houston, TX 77005, USA.
Nanomaterials (Basel). 2024 Sep 29;14(19):1576. doi: 10.3390/nano14191576.
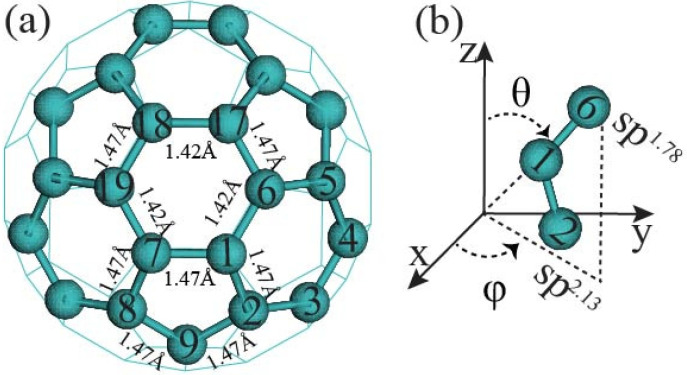
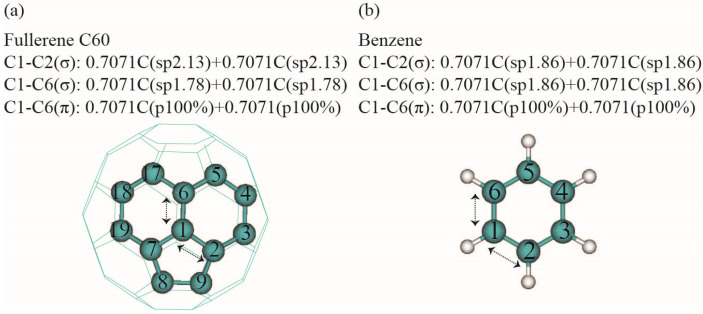
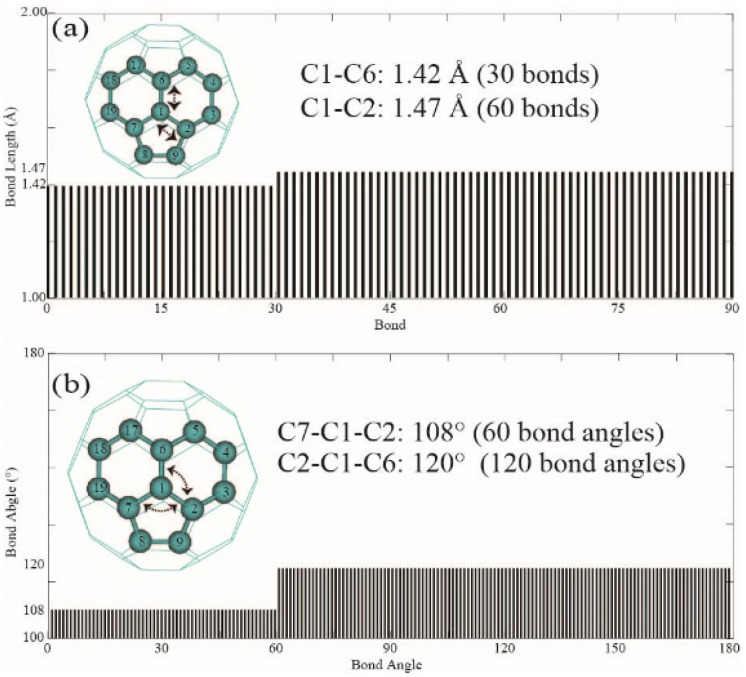
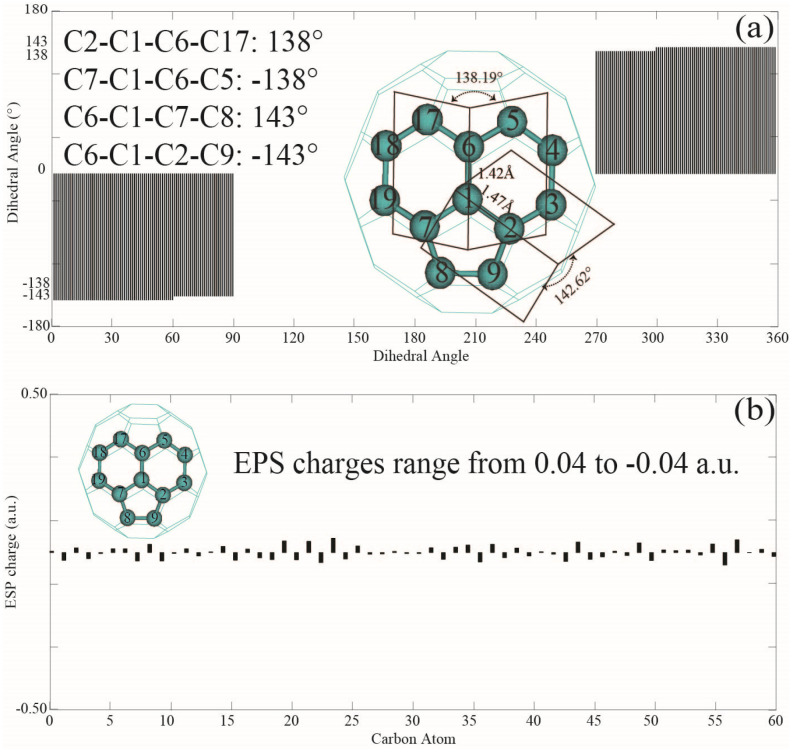
Among C's diverse functionalities, its potential application in CO sequestration has gained increasing interest. However, the processes involved are sensitive to the molecule's electronic structure, aspects of which remain debated and require greater precision. To address this, we performed structural optimization of fullerene C using the QM MP2/6-31G* method. The nonplanarity of the optimized icosahedron is characterized by two types of dihedral angles: 138° and 143°. The 120 dihedrals of 138° occur between two hexagons intersecting at C-C bonds of 1.42 Å, while the 60 dihedrals of 143° are observed between hexagons and pentagons at C-C bonds of 1.47 Å. NBO analysis reveals less pyramidal sp hybridization for carbons at the 1.42 Å bonds and more pyramidal sp hybridization for the 1.47 Å bonds. Electrostatic potential charges range from -0.04 a.u. to 0.04 a.u. on the carbon atoms. Second-order perturbation analysis indicates that delocalization interactions in the C-C bonds of 1.42 Å (143.70 kcal/mol) and 1.47 Å (34.98 kcal/mol) are 22% and 38% higher, respectively, than those in benzene. MP2/Def2SVP calculations yield a correlation energy of 13.49 kcal/mol per electron for C, slightly higher than the 11.68 kcal/mol for benzene. However, the results from HOMO-LUMO calculations should be interpreted with caution. This study may assist in the rational design of fullerene C derivatives for CO reduction systems.
在C的多种功能中,其在二氧化碳封存方面的潜在应用越来越受到关注。然而,所涉及的过程对该分子的电子结构敏感,其中一些方面仍存在争议,需要更高的精度。为了解决这个问题,我们使用量子力学MP2/6-31G*方法对富勒烯C进行了结构优化。优化后的二十面体的非平面性由两种二面角表征:138°和143°。138°的120个二面角出现在以1.42 Å的C-C键相交的两个六边形之间,而143°的60个二面角出现在以1.47 Å的C-C键相连的六边形和五边形之间。自然键轨道(NBO)分析表明,1.42 Å键处的碳原子的sp杂化呈较少的棱锥形,而1.47 Å键处的碳原子的sp杂化呈较多的棱锥形。碳原子上的静电势电荷范围为-0.04 a.u.至0.04 a.u.。二阶微扰分析表明,1.42 Å(143.70 kcal/mol)和1.47 Å(34.98 kcal/mol)的C-C键中的离域相互作用分别比苯中的高22%和38%。MP2/Def2SVP计算得出C的每个电子的相关能为13.49 kcal/mol,略高于苯的11.68 kcal/mol。然而,最高已占分子轨道-最低未占分子轨道(HOMO-LUMO)计算的结果应谨慎解释。本研究可能有助于合理设计用于二氧化碳还原系统的富勒烯C衍生物。