Mammadova Dilbar, Kraus Cornelia, Leis Thomas, Popp Bernt, Zweier Christiane, Reis Andre, Trollmann Regina
Department of Pediatrics, Pediatric Neurology, Friedrich-Alexander Universität Erlangen-Nürnberg, Erlangen, Germany.
Institute of Human Genetics, Friedrich-Alexander Universität Erlangen-Nürnberg, Erlangen, Germany.
Front Neurol. 2024 Oct 2;15:1458109. doi: 10.3389/fneur.2024.1458109. eCollection 2024.
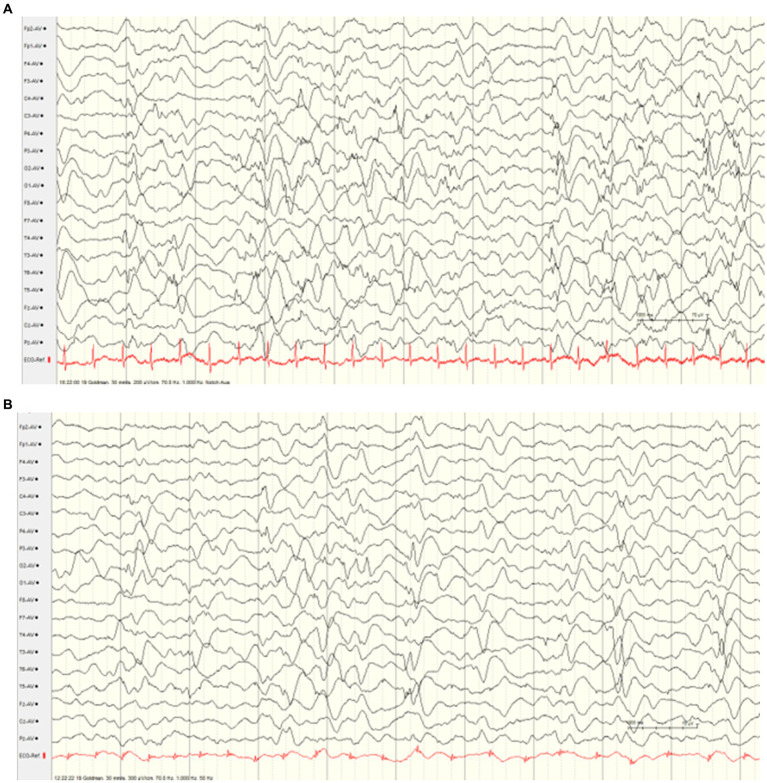
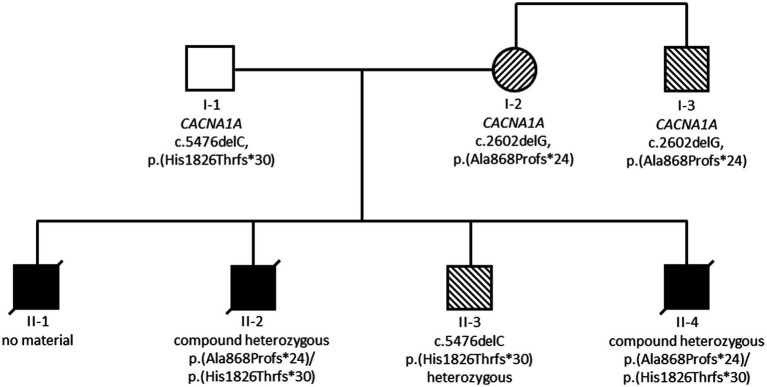
Pathogenic heterozygous variants in are associated with familial hemiplegic migraine, episodic ataxia type 2 and spinocerebellar ataxia type 6, and more recently, neurodevelopmental disorders. We describe a severe, early-onset phenotype including severe muscular hypotonia, early-onset epileptic seizures, apnoea, optic atrophy and dysphagia in three siblings carrying compound heterozygous frameshift variants in . Two male patients died at the age of 5 or 14 months of suspected SIDS or severe developmental epileptic encephalopathy (DEE) with refractory seizures and apnoea. A male child (index patient) developed severe early-onset DEE including seizures and ictal apnoea at the age of 4 weeks. Another male child developed generalized epilepsy and mild intellectual impairment in late infancy, and his mother and his maternal uncle were identified as carriers of a known pathogenic variant [c.2602delG heterozygous, p. (Ala868Profs24)] with a diagnosis of episodic ataxia type 2. This maternal pathogenic variant c.2602delG was detected in the index patient and child 2. Trio-Exome sequencing identified an additional heterozygous pathogenic variant in the gene, c.5476delC, p.(His1826Thrfs30) in the index patient and child 2, which was inherited from the asymptomatic father. In conclusion, the novel compound heterozygosity for two frameshift pathogenic variants, maternally [c.2602delG, p.(Ala868Profs24)] and paternally [c.5476delC, p.(His1826Thrfs3)] is associated with a severe phenotype of early-onset DEE. This observation highlights the necessity of additional analyses to clarify unusual phenotypes even if a pathogenic variant has already been identified, and expands the clinical spectrum of -related disorders.
基因中的致病性杂合变异与家族性偏瘫性偏头痛、发作性共济失调2型和脊髓小脑共济失调6型有关,最近还与神经发育障碍有关。我们描述了一种严重的早发型表型,包括三名携带该基因复合杂合移码变异的兄弟姐妹出现严重肌肉张力减退、早发性癫痫发作、呼吸暂停、视神经萎缩和吞咽困难。两名男性患者分别在5个月或14个月时死于疑似婴儿猝死综合征或伴有难治性癫痫发作和呼吸暂停的严重发育性癫痫性脑病(DEE)。一名男童(索引患者)在4周龄时出现严重的早发性DEE,包括癫痫发作和发作期呼吸暂停。另一名男童在婴儿晚期出现全身性癫痫和轻度智力障碍,他的母亲和舅舅被确定为已知致病性变异[c.2602delG杂合,p.(Ala868Profs24)]的携带者,诊断为发作性共济失调2型。在索引患者和儿童2中检测到这种母源致病性变异c.2602delG。三联外显子组测序在索引患者和儿童2中发现该基因的另一个杂合致病性变异,c.5476delC,p.(His1826Thrfs30),该变异遗传自无症状的父亲。总之,母源[c.2602delG,p.(Ala868Profs24)]和父源[c.5476delC,p.(His1826Thrfs3)]两个移码致病性变异的新型复合杂合性与早发性DEE的严重表型有关。这一观察结果强调,即使已经鉴定出致病性变异,仍需进行额外分析以阐明不寻常的表型,并扩展了该基因相关疾病的临床谱。