Laboratory of Molecular Parasitology, Institut de Biologie et de Médecine Moléculaires (IBMM), Université Libre de Bruxelles, 6041 Gosselies, Belgium.
Cells. 2024 Oct 20;13(20):1738. doi: 10.3390/cells13201738.
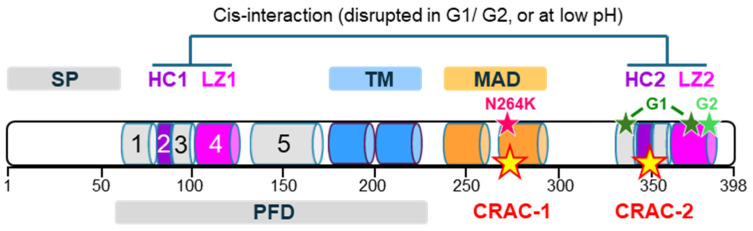
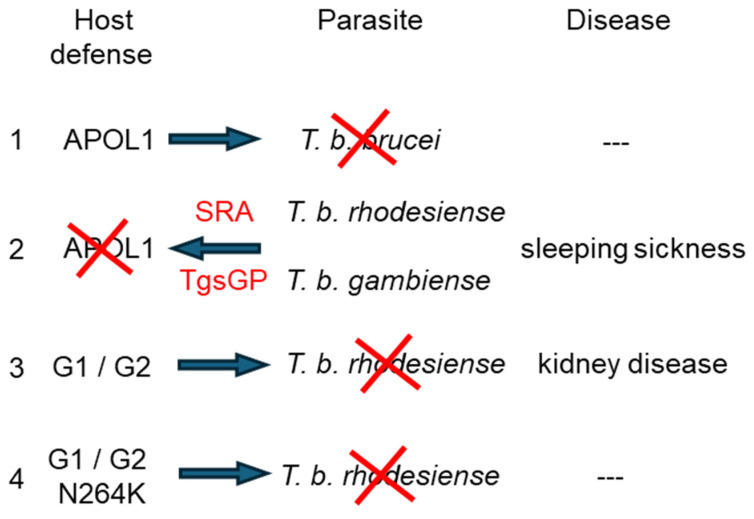
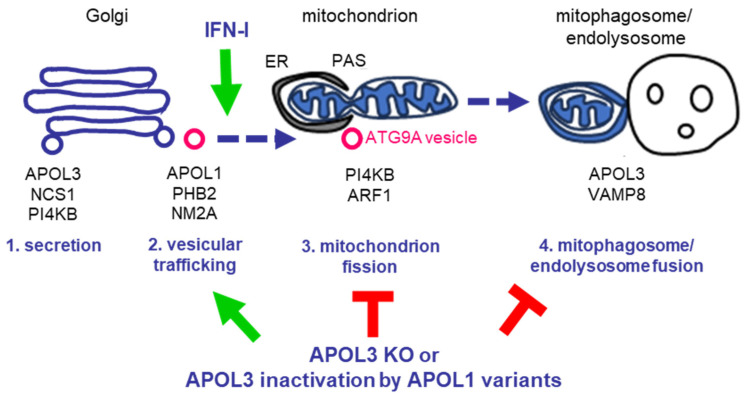
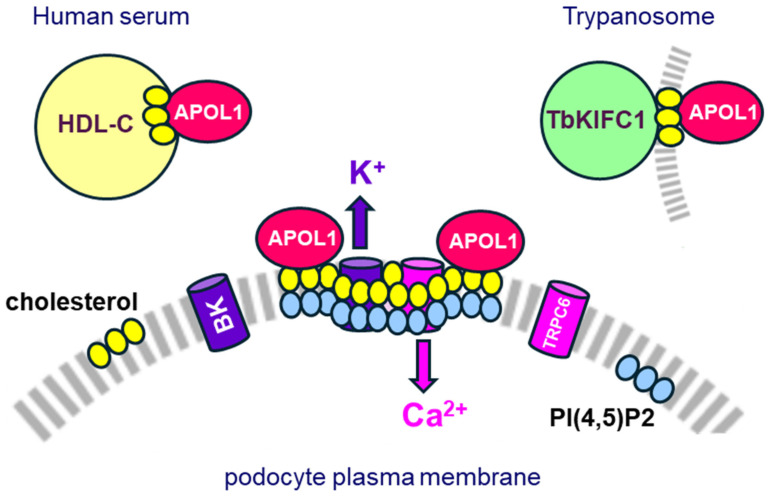
Apolipoprotein-L1 (APOL1) is a membrane-interacting protein induced by inflammation, which confers human resistance to infection by African trypanosomes. APOL1 kills through induction of apoptotic-like parasite death, but two clones acquired resistance to APOL1, allowing them to cause sleeping sickness. An APOL1 C-terminal sequence alteration, such as occurs in natural West African variants G1 and G2, restored human resistance to these clones. However, APOL1 unfolding induced by G1 or G2 mutations enhances protein hydrophobicity, resulting in kidney podocyte dysfunctions affecting renal filtration. The mechanism involved in these dysfunctions is debated. The ability of APOL1 to generate ion pores in trypanosome intracellular membranes or in synthetic membranes was provided as an explanation. However, transmembrane insertion of APOL1 strictly depends on acidic conditions, and podocyte cytopathology mainly results from secreted APOL1 activity on the plasma membrane, which occurs under non-acidic conditions. In this review, I argue that besides inactivation of APOL3 functions in membrane dynamics (fission and fusion), APOL1 variants induce inflammation-linked podocyte toxicity not through pore formation, but through plasma membrane disturbance resulting from increased interaction with cholesterol, which enhances cation channels activity. A natural mutation in the membrane-interacting domain (N264K) abrogates variant APOL1 toxicity at the expense of slightly increased sensitivity to trypanosomes, further illustrating the continuous mutual adaptation between host and parasite.
载脂蛋白 L1(APOL1)是一种由炎症诱导的膜结合蛋白,它赋予人类抵抗非洲锥虫感染的能力。APOL1 通过诱导类似凋亡的寄生虫死亡来杀死寄生虫,但有两个 克隆体对 APOL1 产生了抗性,从而导致昏睡病。APOL1 C 端序列的改变,如在天然的西非变体 G1 和 G2 中发生的改变,恢复了人类对这些克隆体的抗性。然而,G1 或 G2 突变诱导的 APOL1 解折叠增强了蛋白质的疏水性,导致影响肾脏过滤功能的足细胞功能障碍。涉及这些功能障碍的机制存在争议。APOL1 在锥虫细胞内膜或合成膜中产生离子孔的能力被认为是一种解释。然而,APOL1 的跨膜插入严格依赖于酸性条件,而足细胞细胞病变主要是由于 APOL1 在质膜上的分泌活性引起的,这种活性发生在非酸性条件下。在这篇综述中,我认为除了 APOL3 功能在膜动态(分裂和融合)中的失活外,APOL1 变体通过形成孔而不是通过增加与胆固醇的相互作用导致炎症相关的足细胞毒性,从而增强阳离子通道的活性。膜相互作用域(N264K)中的天然突变以牺牲对锥虫的略微增加的敏感性为代价,消除了变体 APOL1 的毒性,进一步说明了宿主和寄生虫之间的持续相互适应。