Talubo Nicholas Dale D, Tsai Po-Wei, Tayo Lemmuel L
School of Chemical, Biological, and Materials Engineering and Sciences, Mapúa University, Manila 1002, Philippines.
School of Graduate Studies, Mapúa University, Manila 1002, Philippines.
Biology (Basel). 2024 Sep 26;13(10):765. doi: 10.3390/biology13100765.
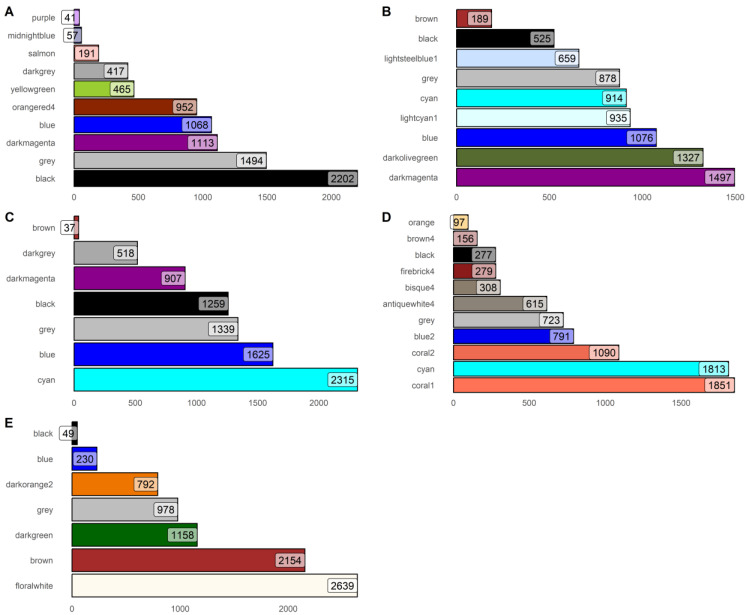
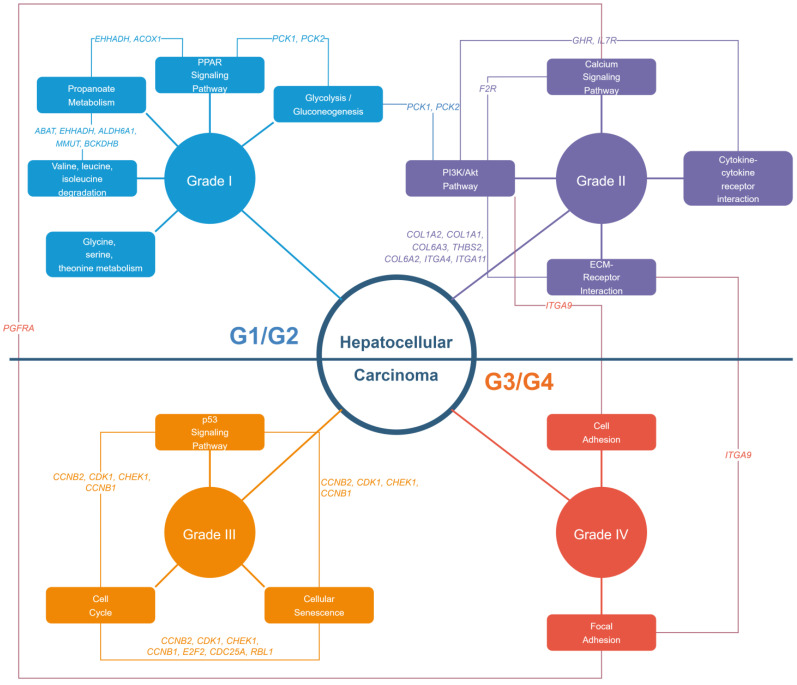
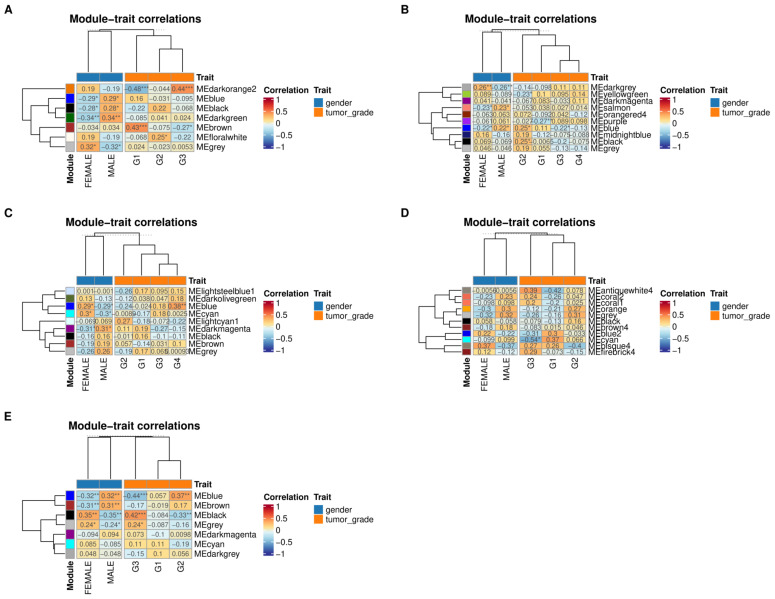
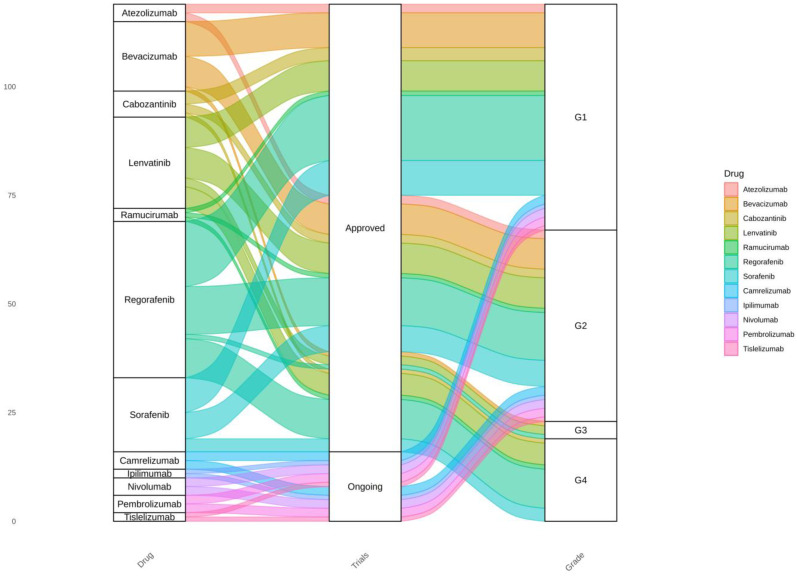
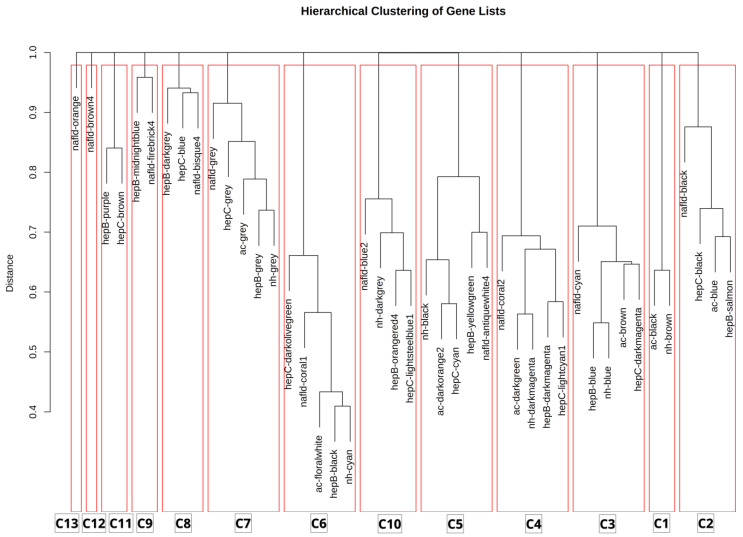
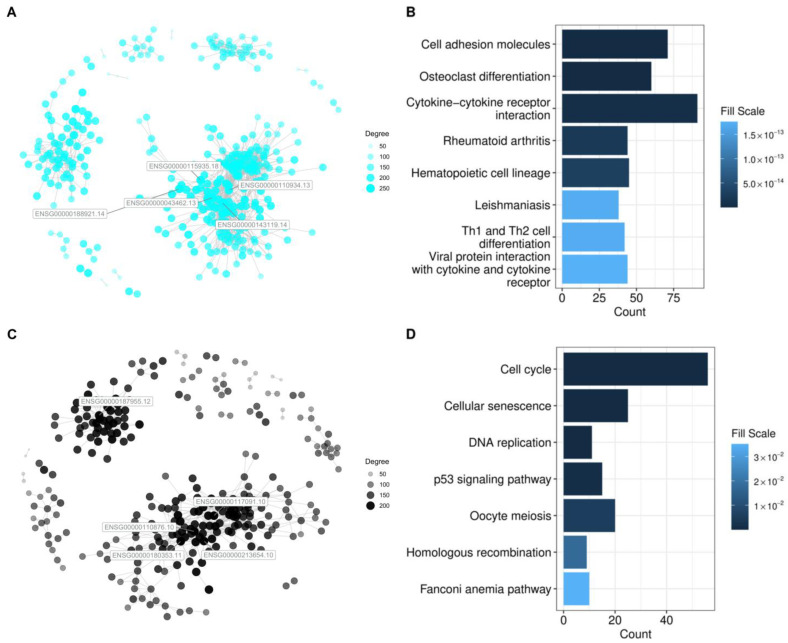
Hepatocellular carcinoma (HCC) has the highest mortality rate and is the most frequent of liver cancers. The heterogeneity of HCC in its etiology and molecular expression increases the difficulty in identifying possible treatments. To elucidate the molecular mechanisms of HCC across grades, data from The Cancer Genome Atlas (TCGA) were used for gene co-expression analysis, categorizing each sample into its pre-existing risk factors. The R library BioNERO was used for preprocessing and gene co-expression network construction. For those modules most correlated with a grade, functional enrichments from different databases were then tested, which appeared to have relatively consistent patterns when grouped by G1/G2 and G3/G4. G1/G2 exhibited the involvement of pathways related to metabolism and the PI3K/Akt pathway, which regulates cell proliferation and related pathways, whereas G3/G4 showed the activation of cell adhesion genes and the p53 signaling pathway, which regulates apoptosis, cell cycle arrest, and similar processes. Module preservation analysis was then used with the no history dataset as the reference network, which found cell adhesion molecules and cell cycle genes to be preserved across all risk factors, suggesting they are imperative in the development of HCC regardless of potential etiology. Through hierarchical clustering, modules related to the cell cycle, cell adhesion, the immune system, and the ribosome were found to be consistently present across all risk factors, with distinct clusters linked to oxidative phosphorylation in viral HCC and pentose and glucuronate interconversions in non-viral HCC, underscoring their potential roles in cancer progression.
肝细胞癌(HCC)的死亡率最高,是最常见的肝癌类型。HCC在病因和分子表达方面的异质性增加了确定可能治疗方法的难度。为了阐明不同分级HCC的分子机制,利用癌症基因组图谱(TCGA)的数据进行基因共表达分析,将每个样本按照其预先存在的风险因素进行分类。使用R语言库BioNERO进行预处理和基因共表达网络构建。对于与分级最相关的那些模块,随后测试了来自不同数据库的功能富集情况,按G1/G2和G3/G4分组时,这些富集情况似乎具有相对一致的模式。G1/G2表现出与代谢和PI3K/Akt信号通路相关的途径的参与,PI3K/Akt信号通路调节细胞增殖及相关途径,而G3/G4显示细胞黏附基因和p53信号通路的激活,p53信号通路调节细胞凋亡、细胞周期阻滞及类似过程。然后以无病史数据集作为参考网络进行模块保留分析,发现细胞黏附分子和细胞周期基因在所有风险因素中均被保留,这表明无论潜在病因如何,它们在HCC的发展中都至关重要。通过层次聚类发现,与细胞周期、细胞黏附、免疫系统和核糖体相关的模块在所有风险因素中均一致存在,在病毒性HCC中,不同簇与氧化磷酸化相关,在非病毒性HCC中与戊糖和葡萄糖醛酸相互转化相关,突出了它们在癌症进展中的潜在作用。