Sabin Susanna J, Beesley Cari A, Marston Chung K, Paisie Taylor K, Gulvik Christopher A, Sprenger Gregory A, Gee Jay E, Traxler Rita M, Bell Melissa E, McQuiston John R, Weiner Zachary P
Laboratory Leadership Service Fellow Assigned to the National Center for Emerging and Zoonotic Infectious Diseases, CDC, Atlanta, GA 30329, USA.
Centers for Disease Control and Prevention, National Center for Emerging and Zoonotic Infectious Diseases, Division of High-Consequence Pathogens and Pathology, Bacterial Special Pathogens Branch, 1600 Clifton Rd, Atlanta, GA 30329, USA.
Pathogens. 2024 Oct 10;13(10):884. doi: 10.3390/pathogens13100884.
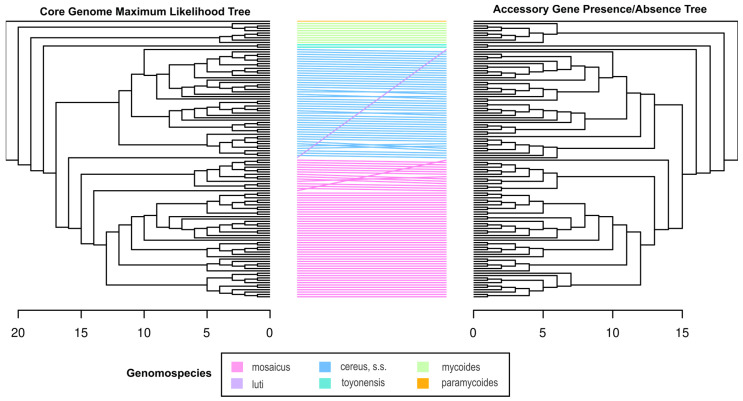
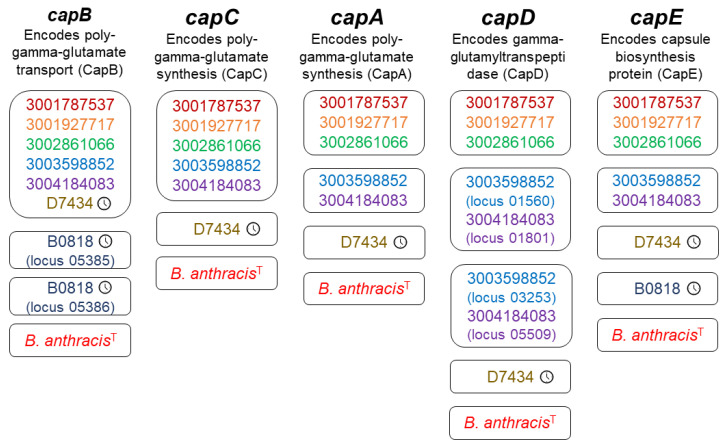
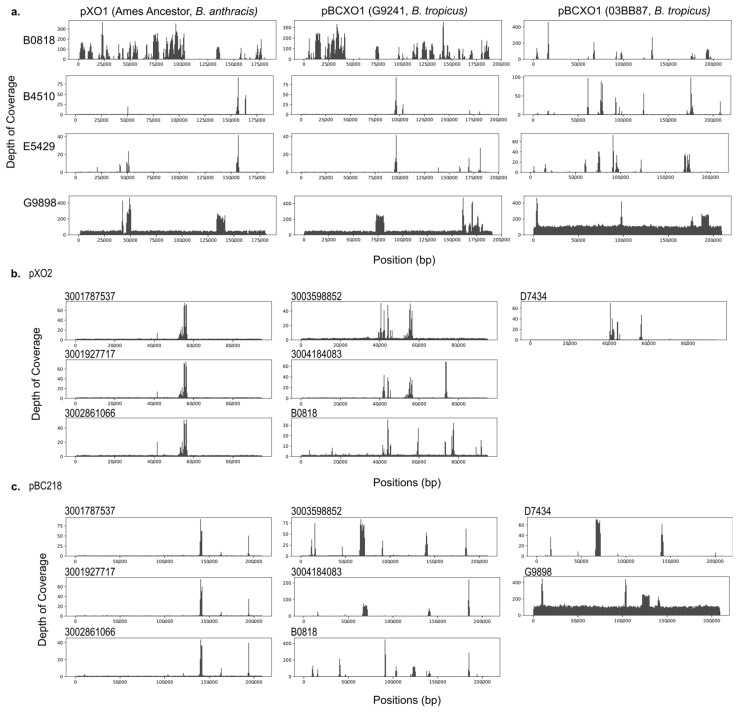
causes anthrax through virulence factors encoded on two plasmids. However, non- organisms within the closely related, environmentally ubiquitous group (BCG) may cause an anthrax-like disease in humans through the partial adoption of anthrax-associated virulence genes, challenging the definition of anthrax disease. To elucidate these phenomena and their evolutionary past, we performed whole-genome sequencing on non- BCG isolates, including 93 archival (1967-2003) and 5 contemporary isolates (2019-2023). We produced annotated genomic assemblies and performed a pan-genome analysis to identify evidence of virulence gene homology and virulence gene acquisition by linear inheritance or horizontal gene transfer. At least one anthrax-associated virulence gene was annotated in ten isolates. Most homologous sequences in archival isolates showed evidence of pseudogenization and subsequent gene loss. The presence or absence of accessory genes, including anthrax-associated virulence genes, aligned with the phylogenetic structure of the BCG core genome. These findings support the hypothesis that anthrax-associated virulence genes were inherited from a common ancestor in the BCG and were retained or lost across different lineages, and contribute to a growing body of work informing public health strategies related to anthrax surveillance and identification.
通过两个质粒上编码的毒力因子引发炭疽病。然而,密切相关的、环境中普遍存在的卡介苗(BCG)组内的非卡介苗生物可能通过部分采用与炭疽相关的毒力基因在人类中引发类似炭疽的疾病,这对炭疽病的定义提出了挑战。为了阐明这些现象及其进化历程,我们对非卡介苗分离株进行了全基因组测序,包括93株存档菌株(1967 - 2003年)和5株当代菌株(2019 - 2023年)。我们生成了注释的基因组组装体,并进行了泛基因组分析,以确定毒力基因同源性以及通过线性遗传或水平基因转移获得毒力基因的证据。在十个分离株中注释到至少一个与炭疽相关的毒力基因。存档菌株中的大多数同源序列显示出假基因化及随后基因丢失的证据。包括与炭疽相关的毒力基因在内的辅助基因的存在与否与卡介苗核心基因组的系统发育结构一致。这些发现支持了这样的假设,即与炭疽相关的毒力基因是从卡介苗的共同祖先遗传而来,并在不同谱系中保留或丢失,并且有助于为与炭疽监测和鉴定相关的公共卫生策略提供信息的研究工作不断增加。