Center for Structural Biology, National Cancer Institute, Frederick, MD 21702, USA.
Department of Molecular Physiology and Biological Physics, University of Virginia, Charlottesville, Virginia, USA.
IUCrJ. 2024 Nov 1;11(Pt 6):966-976. doi: 10.1107/S2052252524009928.
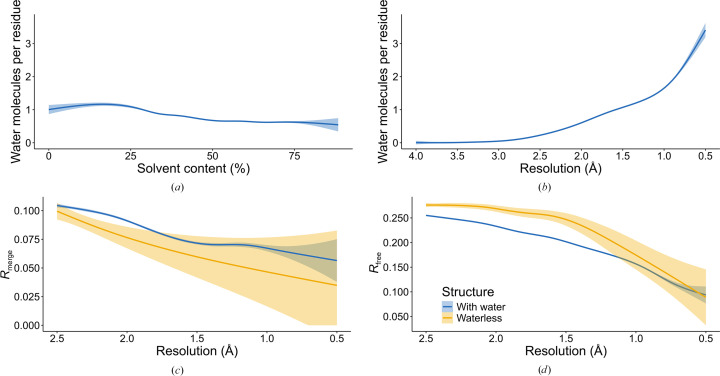
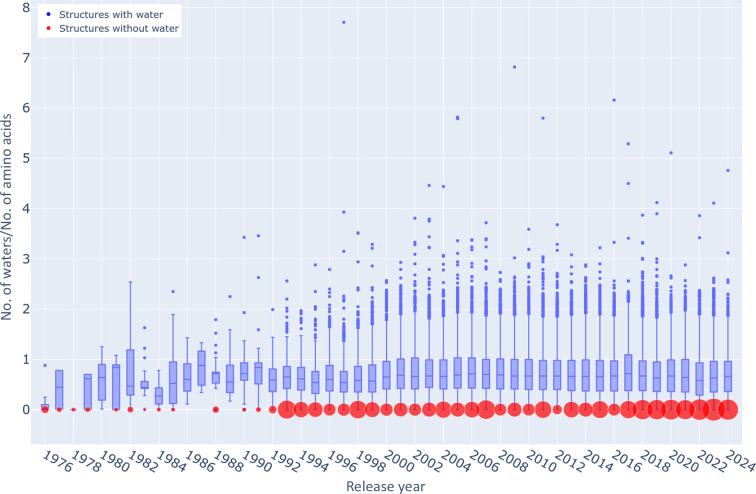
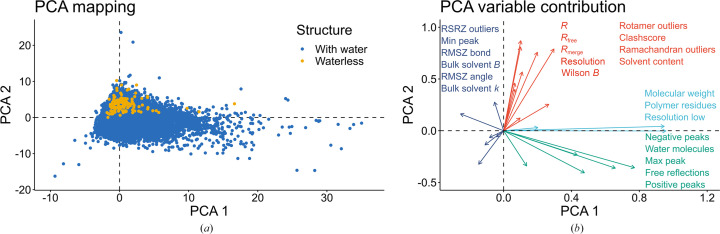
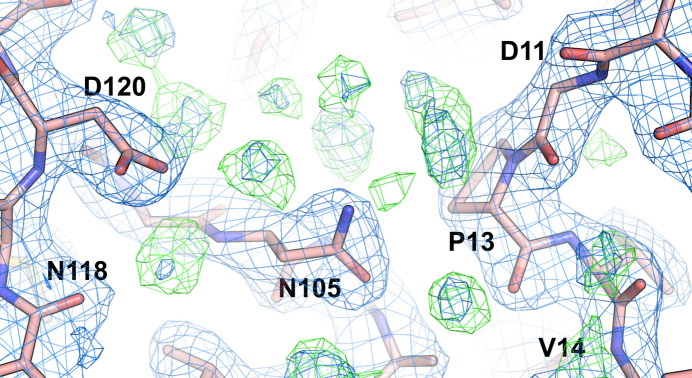
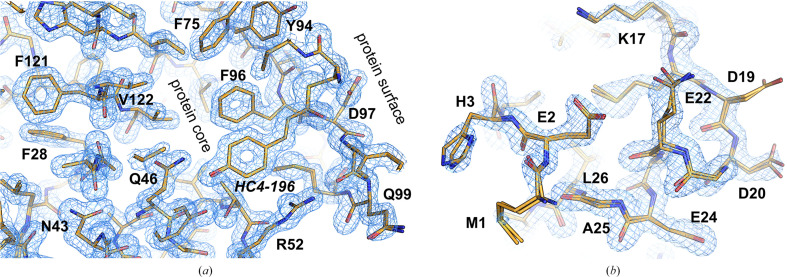
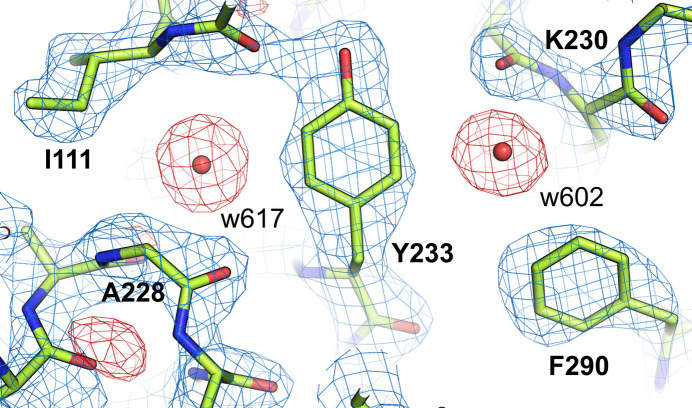
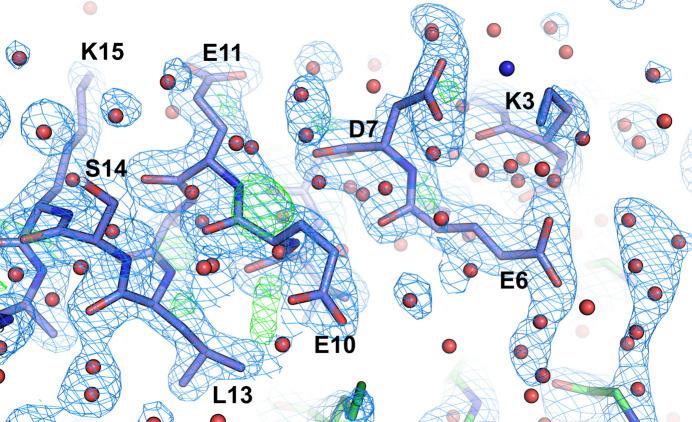
The absence of solvent molecules in high-resolution protein crystal structure models deposited in the Protein Data Bank (PDB) contradicts the fact that, for proteins crystallized from aqueous media, water molecules are always expected to bind to the protein surface, as well as to some sites in the protein interior. An analysis of the contents of the PDB indicated that the expected ratio of the number of water molecules to the number of amino-acid residues exceeds 1.5 in atomic resolution structures, decreasing to 0.25 at around 2.5 Å resolution. Nevertheless, almost 800 protein crystal structures determined at a resolution of 2.5 Å or higher are found in the current release of the PDB without any water molecules, whereas some other depositions have unusually low or high occupancies of modeled solvent. Detailed analysis of these depositions revealed that the lack of solvent molecules might be an indication of problems with either the diffraction data, the refinement protocol, the deposition process or a combination of these factors. It is postulated that problems with solvent structure should be flagged by the PDB and addressed by the depositors.
在蛋白质数据库(PDB)中储存的高分辨率蛋白质晶体结构模型中没有溶剂分子,这与以下事实相矛盾:对于从水介质中结晶的蛋白质,水分子总是预期与蛋白质表面以及蛋白质内部的某些部位结合。对 PDB 内容的分析表明,在原子分辨率结构中,水分子与氨基酸残基数的预期比值超过 1.5,在分辨率约为 2.5 Å 时降至 0.25。然而,目前 PDB 版本中发现了近 800 个在 2.5 Å 或更高分辨率下确定的蛋白质晶体结构,没有任何水分子,而其他一些结构的模型溶剂占有率异常低或高。对这些结构的详细分析表明,缺乏溶剂分子可能表明衍射数据、精修方案、沉积过程或这些因素的组合存在问题。有人假设,溶剂结构的问题应该由 PDB 标记,并由存款人解决。