Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA.
Genes & Human Disease Research Program, Oklahoma Medical Research Foundation, Oklahoma City, OK 73104, USA.
Am J Hum Genet. 2024 Nov 7;111(11):2566-2581. doi: 10.1016/j.ajhg.2024.10.002. Epub 2024 Oct 28.
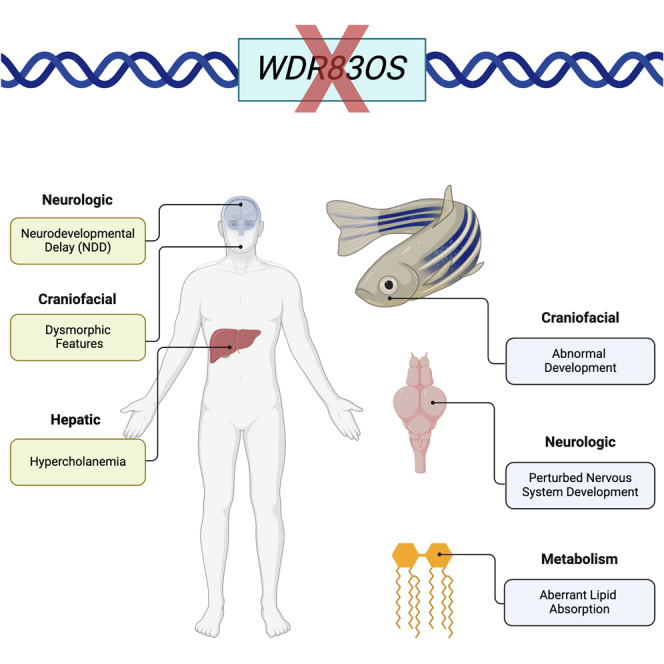
WD repeat domain 83 opposite strand (WDR83OS) encodes the 106-aa (amino acid) protein Asterix, which heterodimerizes with CCDC47 to form the PAT (protein associated with ER translocon) complex. This complex functions as a chaperone for large proteins containing transmembrane domains to ensure proper folding. Until recently, little was known about the role of WDR83OS or CCDC47 in human disease traits. However, biallelic variants in CCDC47 were identified in four unrelated families with trichohepatoneurodevelopmental syndrome, characterized by a neurodevelopmental disorder (NDD) with liver dysfunction. Three affected siblings in an additional family share a homozygous truncating WDR83OS variant and a phenotype of NDD, dysmorphic features, and liver dysfunction. Using family-based rare variant analyses of exome sequencing (ES) data and case matching through GeneMatcher, we describe the clinical phenotypes of 11 additional individuals in eight unrelated families (nine unrelated families, 14 individuals in total) with biallelic putative truncating variants in WDR83OS. Consistent clinical features include NDD (14/14), facial dysmorphism (13/14), intractable itching (9/14), and elevated bile acids (5/6). Whereas bile acids were significantly elevated in 5/6 of individuals tested, bilirubin was normal and liver enzymes were normal to mildly elevated in all 14 individuals. In three of six individuals for whom longitudinal data were available, we observed a progressive reduction in relative head circumference. A zebrafish model lacking Wdr83os function further supports its role in the nervous system, craniofacial development, and lipid absorption. Taken together, our data support a disease-gene association between biallelic loss-of-function of WDR83OS and a neurological disease trait with hypercholanemia.
WD 重复结构域 83 反义链(WDR83OS)编码 106 个氨基酸的 Asterix 蛋白,该蛋白与 CCDC47 异二聚形成 PAT(与内质网转位复合物相关的蛋白)复合物。该复合物作为含有跨膜结构域的大蛋白的伴侣,确保其正确折叠。直到最近,人们对 WDR83OS 或 CCDC47 在人类疾病特征中的作用知之甚少。然而,在四个无关联的家族中发现了 CCDC47 的双等位基因变异,这些家族患有毛发-肝-神经发育不良综合征,其特征为神经发育障碍(NDD)伴肝功能障碍。另外一个家族的三个受影响的兄弟姐妹携带纯合截断 WDR83OS 变异体和 NDD、畸形特征和肝功能障碍的表型。我们通过外显子组测序(ES)数据的基于家系的罕见变异分析和通过 GeneMatcher 的病例匹配,描述了 8 个无关联家族中 11 个额外个体(9 个无关联家族,共 14 人)的临床表型,这些个体携带 WDR83OS 中的双等位基因推定截断变异体。一致的临床特征包括 NDD(14/14)、面部畸形(13/14)、顽固性瘙痒(9/14)和胆汁酸升高(5/6)。虽然在 5/6 个受检个体中胆汁酸显著升高,但胆红素正常,所有 14 个个体的肝酶正常或轻度升高。在 6 个有纵向数据的个体中,我们观察到相对头围逐渐缩小。缺乏 Wdr83os 功能的斑马鱼模型进一步支持了其在神经系统、颅面发育和脂质吸收中的作用。总之,我们的数据支持 WDR83OS 双等位基因功能丧失与高胆汁血症的神经疾病特征之间存在疾病-基因关联。