Department of Molecular Medicine and Surgery, Karolinska Institutet, 171 76 Stockholm, Sweden.
Department of Clinical Genetics and Genomics, Karolinska University Hospital, 171 77 Stockholm, Sweden.
Genome Res. 2024 Nov 20;34(11):1763-1773. doi: 10.1101/gr.279263.124.
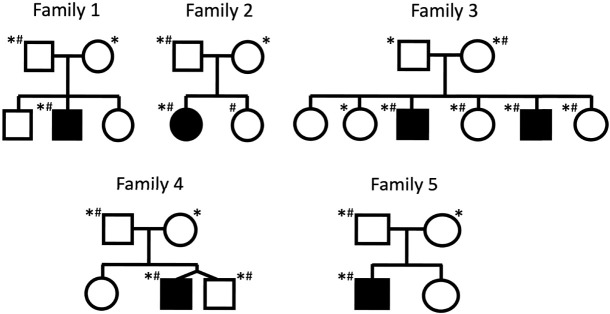
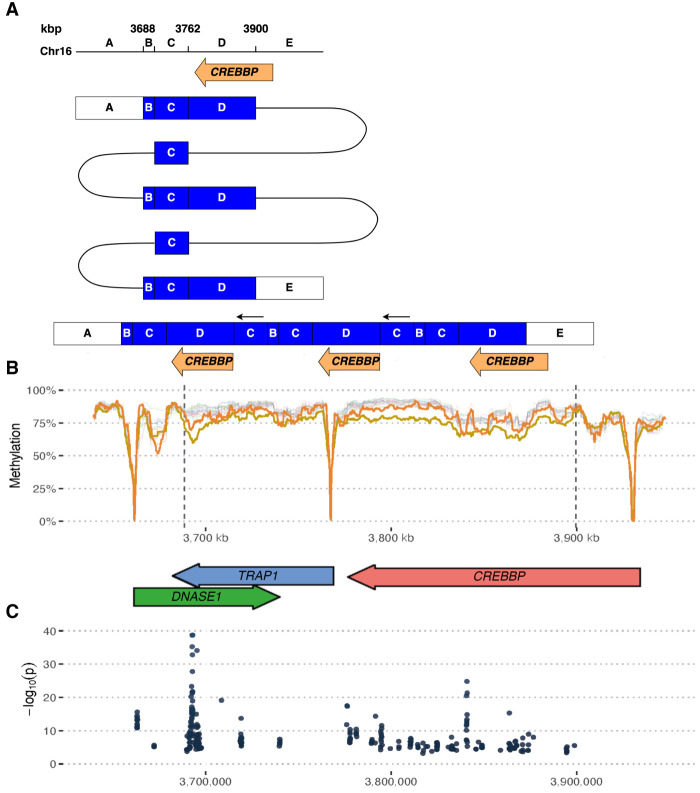
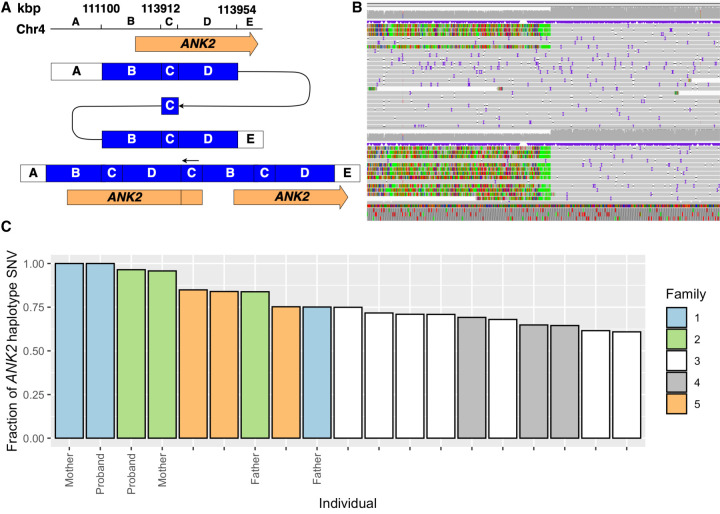
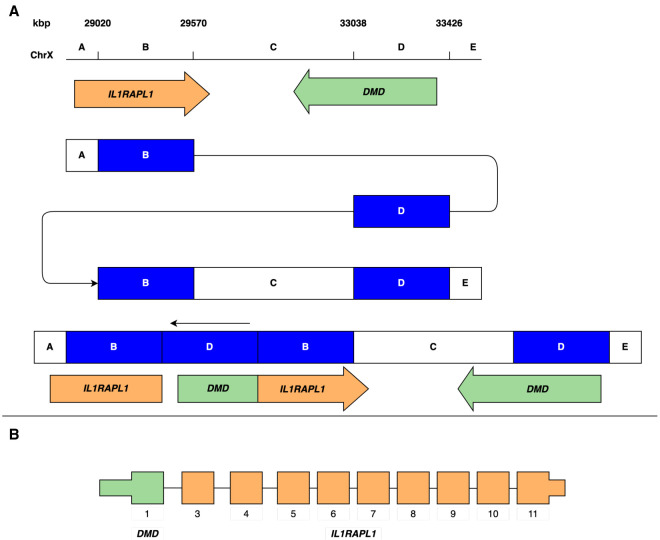
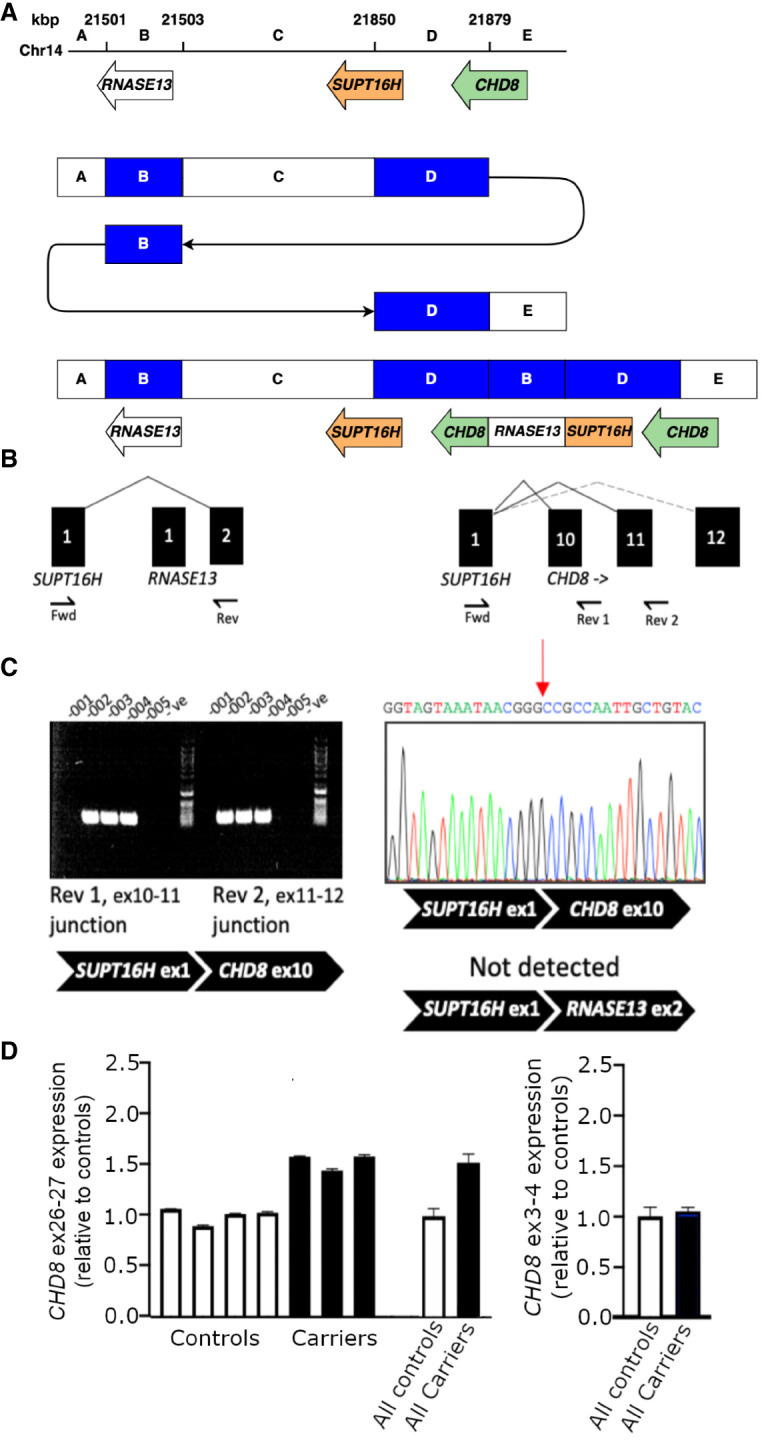
Rare or de novo structural variation, primarily in the form of copy number variants, is detected in 5%-10% of autism spectrum disorder (ASD) families. While complex structural variants involving duplications can generally be detected using microarray or short-read genome sequencing (GS), these methods frequently fail to characterize breakpoints at nucleotide resolution, requiring additional molecular methods for validation and fine-mapping. Here, we use Oxford Nanopore Technologies PromethION long-read GS to characterize complex genomic rearrangements (CGRs) involving large duplications that segregate with ASD in five families. In total, we investigated 13 CGR carriers and were able to resolve all breakpoint junctions at nucleotide resolution. While all breakpoints were identified, the precise genomic architecture of one rearrangement remained unresolved with three different potential structures. The findings in two families include potential fusion genes formed through duplication rearrangements, involving and In two of the families originating from the same geographical region, an identical rearrangement involving was identified, which likely represents a founder variant. In addition, we analyze methylation status directly from the long-read data, allowing us to assess the activity of rearranged genes and regulatory regions. Investigation of methylation across the CGRs reveals aberrant methylation status in carriers across a rearrangement affecting the locus. In aggregate, our results demonstrate the utility of nanopore sequencing to pinpoint CGRs associated with ASD in five unrelated families, and highlight the importance of a gene-centric description of disease-associated complex chromosomal rearrangements.
在 5%-10%的自闭症谱系障碍 (ASD) 家庭中,检测到罕见或新生的结构变异,主要表现为拷贝数变异。虽然涉及重复的复杂结构变异通常可以使用微阵列或短读长基因组测序 (GS) 来检测,但这些方法通常无法在核苷酸分辨率下表征断点,需要额外的分子方法进行验证和精细映射。在这里,我们使用 Oxford Nanopore Technologies PromethION 长读长 GS 来表征与 ASD 共分离的五个家庭中的复杂基因组重排 (CGR),这些重排涉及大的重复。总共,我们研究了 13 个 CGR 携带者,并能够以核苷酸分辨率解析所有断点连接处。虽然所有的断点都被识别出来,但一个重排的精确基因组结构仍然没有得到解决,有三种不同的潜在结构。两个家庭的发现包括通过重复重排形成的潜在融合基因,涉及 和 。在两个来自同一地理区域的家庭中,鉴定到涉及 的相同重排,这可能代表一个起始变体。此外,我们直接从长读长数据中分析甲基化状态,从而能够评估重排基因和调控区域的活性。对 CGR 进行甲基化分析显示,在影响 基因座的重排携带者中存在异常甲基化状态。总的来说,我们的结果表明纳米孔测序在五个不相关的家庭中确定与 ASD 相关的 CGR 的实用性,并强调了对疾病相关复杂染色体重排进行基因中心描述的重要性。