Makino Junichiro, Ebisuzaki Toshikazu, Himeno Ryutaro, Hayashizaki Yoshihide
Advanced Accelerating Systems Co. Ltd, Deiki 1-28, B1312, Kanazawa-ku, Yokohama, Kanagawa, 236-0021, Japan.
Department of Planetology, Graduate School of Science, Kobe University, 1-1, Rokkodai-cho, Nada-ku, Kobe, 657-8051, Japan.
Genomics Inform. 2024 Oct 29;22(1):19. doi: 10.1186/s44342-024-00012-5.
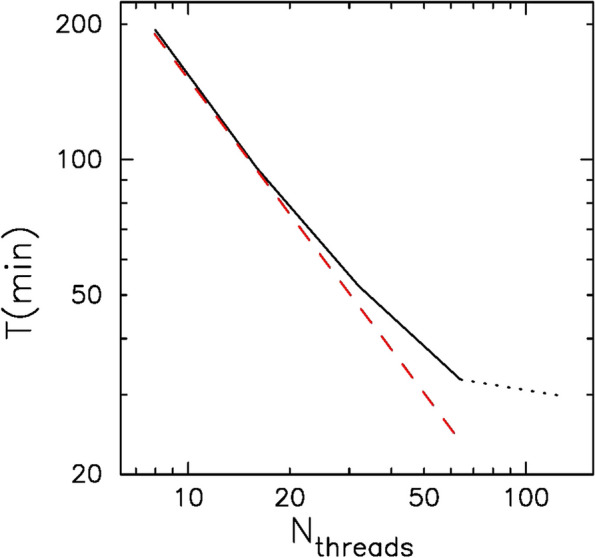
Rapidly increasing the amount of short-read data generated by NGSs (new-generation sequencers) calls for the development of fast and accurate read alignment programs. The programs based on the hash table (BLAST) and Burrows-Wheeler transform (bwa-mem) are used, and the latter is known to give superior performance. We here present a new algorithm, a hybrid of hash table and suffix tree, which we designed to speed up the alignment of short reads against large reference sequences such as the human genome. The total turnaround time for processing one human genome sample (read depth of 30) is just 31 min with our system while that was more than 25 h with bwa-mem/gatk. The time for the aligner alone is 28 min for our system but around 2 h for bwa-mem. Our new algorithm is 4.4 times faster than bwa-mem while achieving similar accuracy. Variant calling and other downstream analyses after the alignment can be done with open-source tools such as SAMtools and Genome Analysis Toolkit (gatk) packages, as well as our own fast variant caller, which is well parallelized and much faster than gatk.
新一代测序仪(NGS)产生的短读长数据量迅速增加,这就需要开发快速且准确的读段比对程序。人们使用了基于哈希表的程序(BLAST)和基于Burrows-Wheeler变换的程序(bwa-mem),后者性能更优。我们在此展示一种新算法,它是哈希表和后缀树的混合算法,旨在加快短读段与大型参考序列(如人类基因组)的比对速度。使用我们的系统处理一个人类基因组样本(读长深度为30)的总周转时间仅为31分钟,而使用bwa-mem/gatk则超过25小时。仅比对程序而言,我们的系统耗时28分钟,而bwa-mem约需2小时。我们的新算法比bwa-mem快4.4倍,同时具有相似的准确性。比对后的变异检测及其他下游分析可使用诸如SAMtools和基因组分析工具包(gatk)软件包等开源工具,以及我们自己开发的快速变异检测程序,该程序具有良好的并行性,比gatk快得多。