College of Engineering, Cornell University, Ithaca, NY, United States of America.
Northwestern-Argonne Institute for Science and Engineering, Evanston, IL, United States of America.
PLoS One. 2024 Oct 30;19(10):e0312686. doi: 10.1371/journal.pone.0312686. eCollection 2024.
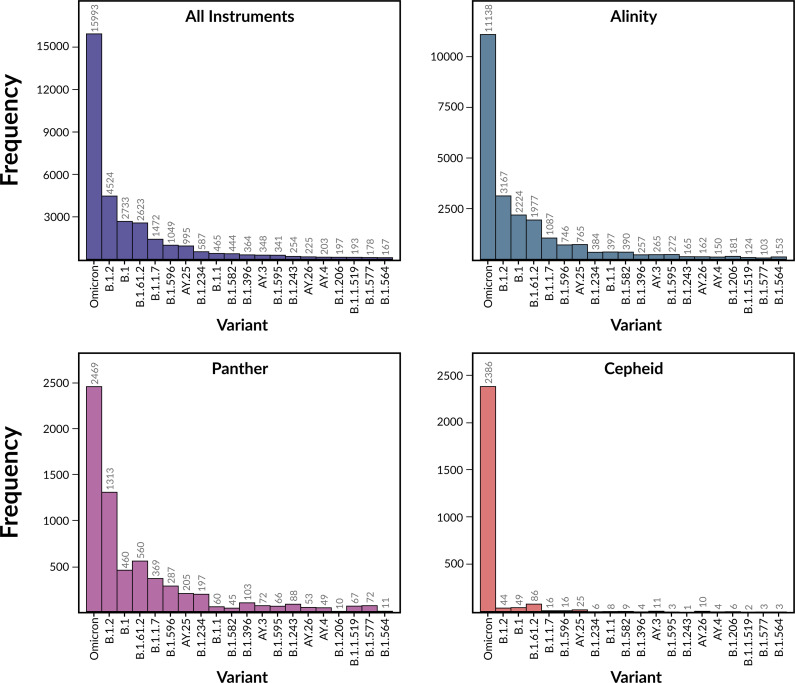
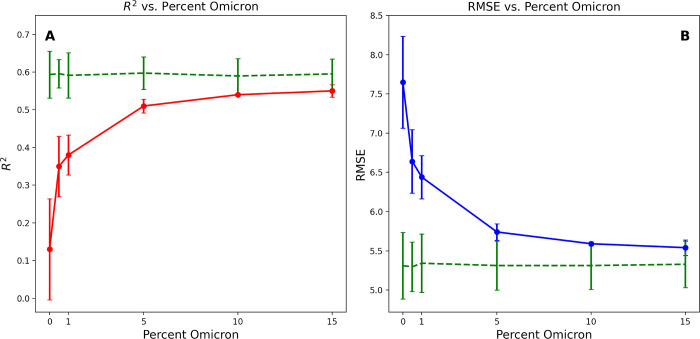
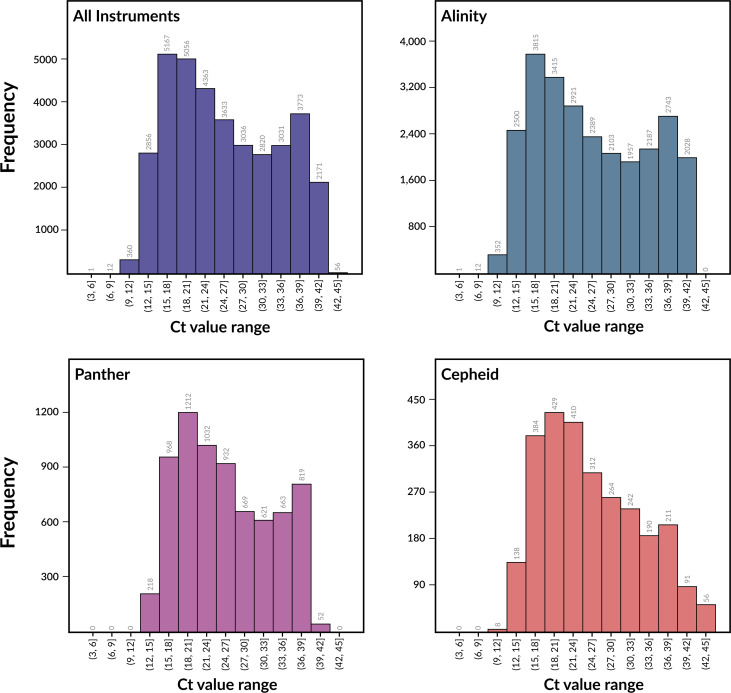
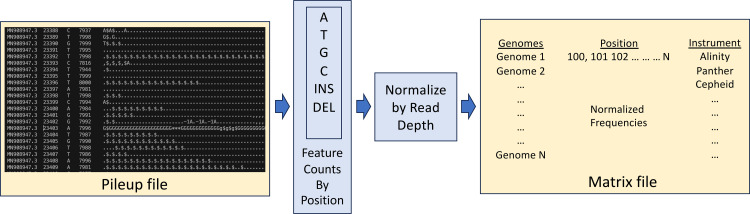
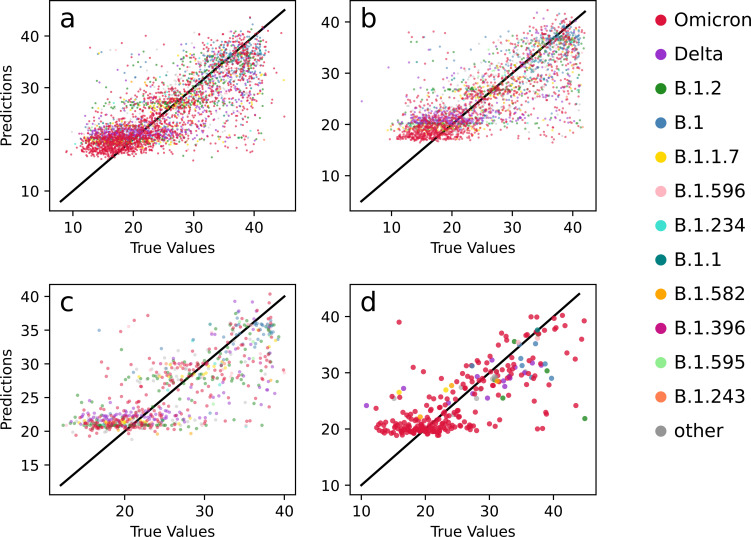
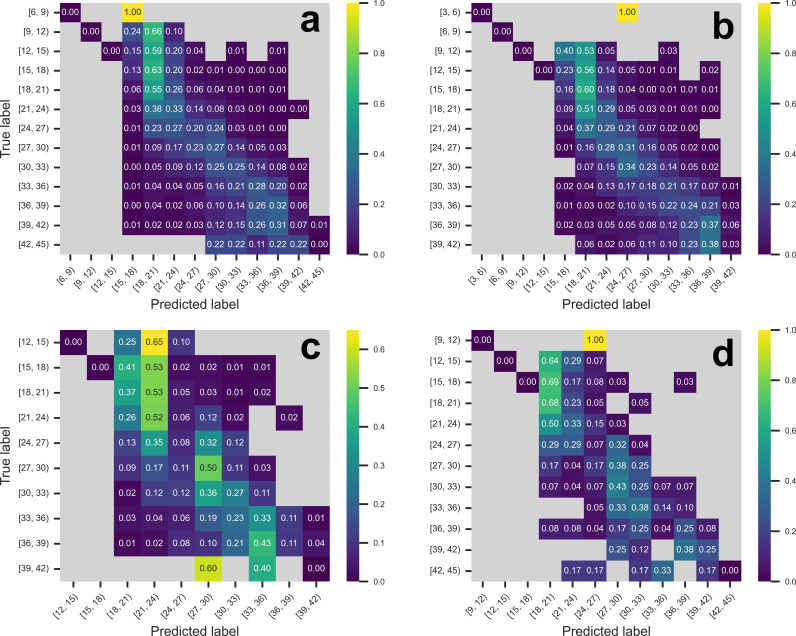
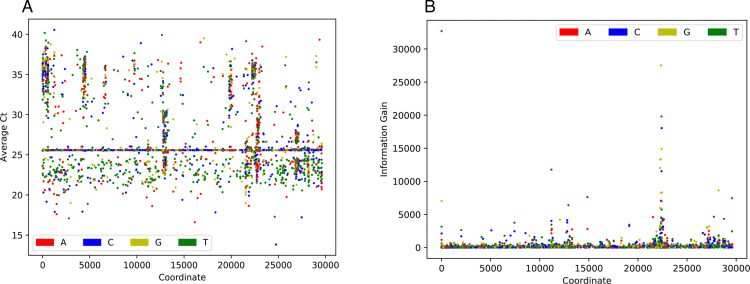
Over the last four years, each successive wave of the COVID-19 pandemic has been caused by variants with mutations that improve the transmissibility of the virus. Despite this, we still lack tools for predicting clinically important features of the virus. In this study, we show that it is possible to predict the PCR cycle threshold (Ct) values from clinical detection assays using sequence data. Ct values often correspond with patient viral load and the epidemiological trajectory of the pandemic. Using a collection of 36,335 high quality genomes, we built models from SARS-CoV-2 intrahost single nucleotide variant (iSNV) data, computing XGBoost models from the frequencies of A, T, G, C, insertions, and deletions at each position relative to the Wuhan-Hu-1 reference genome. Our best model had an R2 of 0.604 [0.593-0.616, 95% confidence interval] and a Root Mean Square Error (RMSE) of 5.247 [5.156-5.337], demonstrating modest predictive power. Overall, we show that the results are stable relative to an external holdout set of genomes selected from SRA and are robust to patient status and the detection instruments that were used. This study highlights the importance of developing modeling strategies that can be applied to publicly available genome sequence data for use in disease prevention and control.
在过去的四年中,COVID-19 大流行的每一波都是由具有提高病毒传播能力的突变的变体引起的。尽管如此,我们仍然缺乏预测病毒临床重要特征的工具。在这项研究中,我们表明,使用序列数据从临床检测分析中预测聚合酶链反应(PCR)循环阈值(Ct)值是可能的。Ct 值通常与患者的病毒载量和大流行的流行病学轨迹相对应。使用 36335 个高质量基因组的集合,我们从 SARS-CoV-2 宿主内单核苷酸变异(iSNV)数据构建了模型,计算了相对于武汉-Hu-1 参考基因组的每个位置的 A、T、G、C、插入和缺失频率的 XGBoost 模型。我们最好的模型的 R2 为 0.604 [0.593-0.616,95%置信区间],均方根误差(RMSE)为 5.247 [5.156-5.337],显示出适度的预测能力。总体而言,我们表明,与从 SRA 中选择的外部保留基因组集相比,结果是稳定的,并且对患者状态和使用的检测仪器具有鲁棒性。这项研究强调了开发可应用于公共基因组序列数据的建模策略的重要性,以用于疾病预防和控制。