Brown Nathaniel K, Shivakumar Vikram S, Langmead Ben
Department of Computer Science, Johns Hopkins University, Baltimore MD 21218.
bioRxiv. 2024 Nov 2:2024.10.29.620953. doi: 10.1101/2024.10.29.620953.
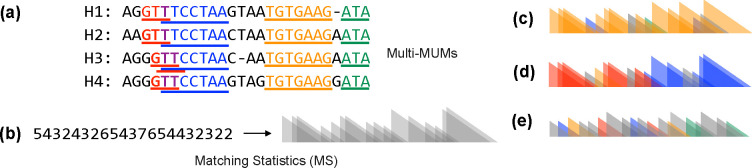
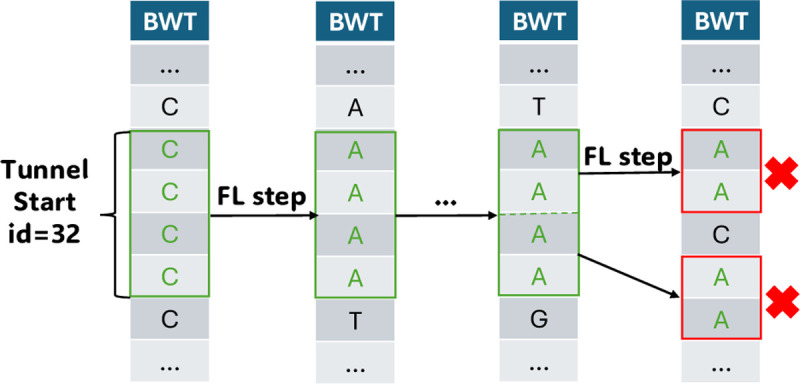
Compressed full-text indexes enable efficient sequence classification against a pangenome or tree-of-life index. Past work on compressed-index classification used matching statistics or pseudo-matching lengths to capture the fine-grained co-linearity of exact matches. But these fail to capture coarse-grained information about whether seeds appear co-linearly in the reference. We present a novel approach that additionally obtains coarse-grained co-linearity ("chain") statistics. We do this without using a chaining algorithm, which would require superlinear time in the number of matches. We start with a collection of strings, avoiding the multiple-alignment step required by graph approaches. We rapidly compute multi-maximal unique matches (multi-MUMs) and identify BWT sub-runs that correspond to these multi-MUMs. From these, we select those that can be "tunneled," and mark these with the corresponding multi-MUM identifiers. This yields an -space index for a collection of sequences having a length- BWT consisting of maximal equal-character runs. Using the index, we simultaneously compute fine-grained matching statistics and coarse-grained chain statistics in linear time with respect to query length. We found that this substantially improves classification accuracy compared to past compressed-indexing approaches and reaches the same level of accuracy as less efficient alignment-based methods.
压缩全文索引能够针对泛基因组或生命树索引进行高效的序列分类。过去关于压缩索引分类的工作使用匹配统计或伪匹配长度来捕获精确匹配的细粒度共线性。但这些方法未能捕获关于种子在参考序列中是否共线出现的粗粒度信息。我们提出了一种新颖的方法,该方法还能获得粗粒度共线性(“链”)统计信息。我们无需使用链接算法来实现这一点,因为链接算法在匹配数量上需要超线性时间。我们从一组字符串开始,避免了图方法所需的多重比对步骤。我们快速计算多最大唯一匹配(multi-MUMs)并识别与这些多最大唯一匹配相对应的BWT子运行。从中,我们选择那些可以“隧穿”的,并使用相应的多最大唯一匹配标识符进行标记。这为长度为 的由 个最大等字符运行组成的BWT的 个序列集合生成了一个 空间索引。使用该索引,我们相对于查询长度在线性时间内同时计算细粒度匹配统计信息和粗粒度链统计信息。我们发现,与过去的压缩索引方法相比,这显著提高了分类准确率,并且达到了与效率较低的基于比对的方法相同的准确率水平。