Stoneman Hayley R, Price Adelle, Gignoux Christopher R, Hendricks Audrey E
Department of Biomedical Informatics, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA.
Human Medical Genetics and Genomics Program, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA.
bioRxiv. 2024 Oct 29:2024.10.24.619530. doi: 10.1101/2024.10.24.619530.
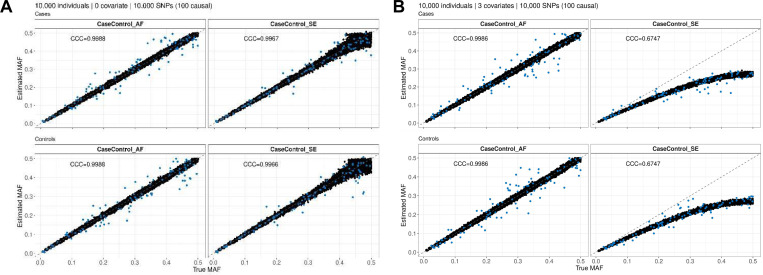
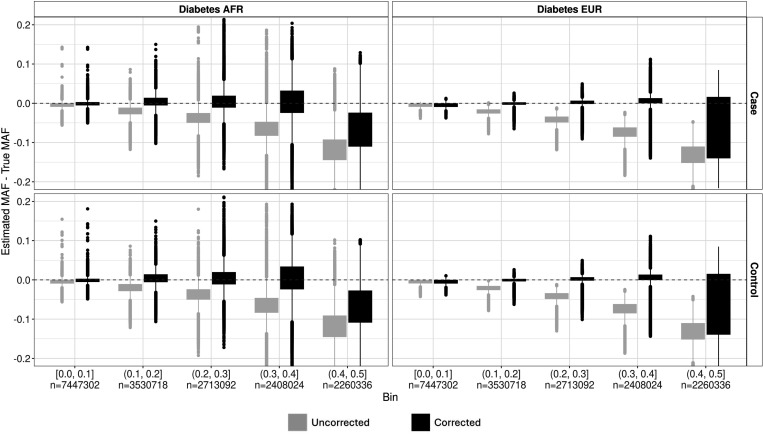
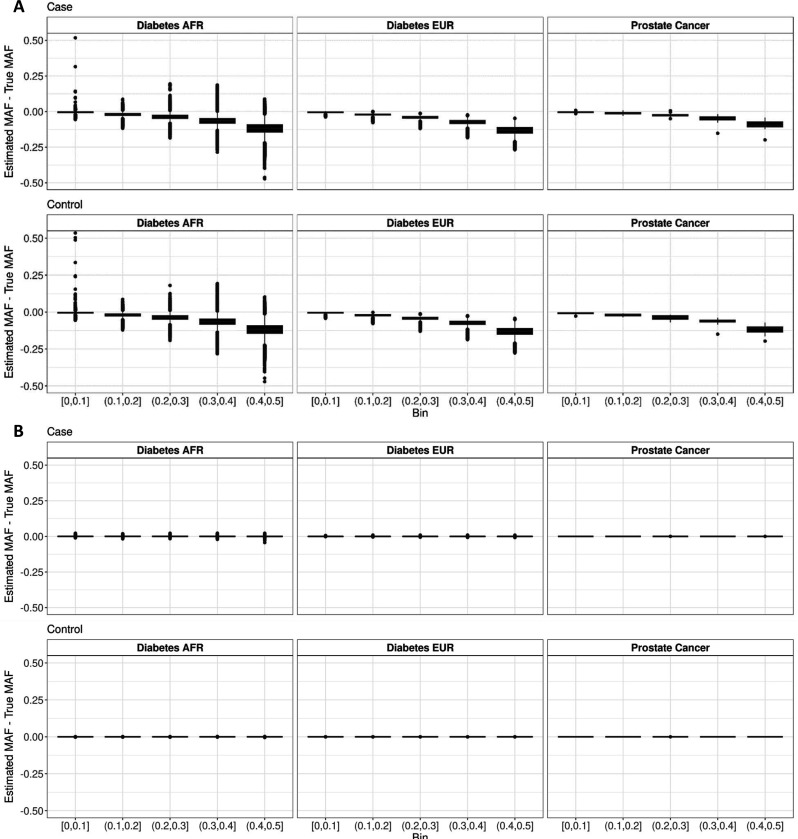
Methods involving summary statistics in genetics can be quite powerful but can be limited in utility. For instance, many post-hoc analyses of disease studies require case and control allele frequencies (AFs), which are not always published. We present two frameworks to derive case and control AFs from GWAS summary statistics using the odds ratio, case and control sample sizes, and either the total (case and control aggregated) AF or standard error (SE). In simulations and real data, derivations of case and controls AFs using total AF is highly accurate across all settings (e.g., minor AF, condition prevalence). Conversely, derivations using SE underestimate common variant AFs (e.g. minor allele frequency >0.3) in the presence of covariates. We develop an adjustment using gnomAD AFs as a proxy for true AFs, which reduces the bias when using SE. While estimating case and control AFs using the total AF is preferred due to its high accuracy, estimating from the SE can be used more broadly since SE can be derived from p-values and beta estimates, which are commonly provided. The methods provided here expand the utility of publicly available genetic summary statistics and promote the reusability of genomic data. The R package with implementations of both methods, is freely available on Bioconductor and GitHub.
遗传学中涉及汇总统计的方法可能非常强大,但实用性可能有限。例如,许多疾病研究的事后分析需要病例组和对照组的等位基因频率(AF),而这些频率并不总是会公布。我们提出了两个框架,可利用优势比、病例组和对照组样本量以及总(病例组和对照组汇总)AF或标准误(SE),从全基因组关联研究(GWAS)汇总统计数据中推导病例组和对照组的AF。在模拟和实际数据中,使用总AF推导病例组和对照组的AF在所有情况下(例如,次要等位基因频率、疾病患病率)都非常准确。相反,在存在协变量的情况下,使用SE进行推导会低估常见变异的AF(例如,次要等位基因频率>0.3)。我们开发了一种使用gnomAD AF作为真实AF的替代指标的调整方法,该方法可减少使用SE时的偏差。虽然由于其高准确性,使用总AF估计病例组和对照组的AF是首选,但由于SE可以从p值和β估计值推导得出(这些通常都会提供),因此从SE进行估计可以更广泛地使用。本文提供的方法扩展了公开可用的遗传汇总统计数据的实用性,并促进了基因组数据的可重用性。包含这两种方法实现的R包可在Bioconductor和GitHub上免费获取。