Sethi Aaftaab, Kumar Janish, Vemula Divya, Gadde Divya, Talla Venu, Qureshi Insaf A, Alvala Mallika
Department of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research (NIPER) Hyderabad 500037 India
Laboratory of Biomolecular Interactions and Transport, Department of Gene Expression, Institute of Molecular Biology and Biotechnology, Faculty of Biology, Adam Mickiewicz University Uniwersytetu Poznanskiego 6 Poznan 61-614 Poland.
RSC Adv. 2024 Nov 18;14(49):36794-36803. doi: 10.1039/d4ra06715k. eCollection 2024 Nov 11.
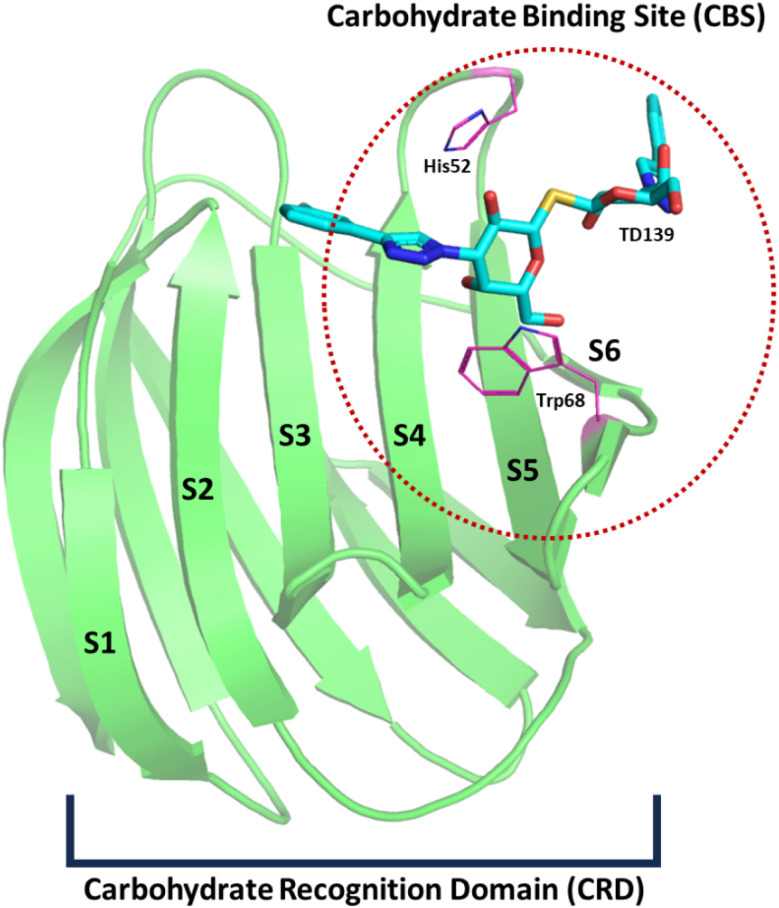
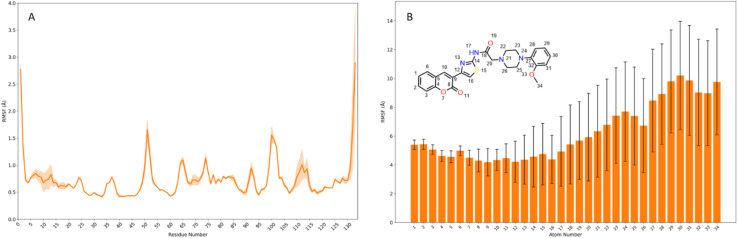
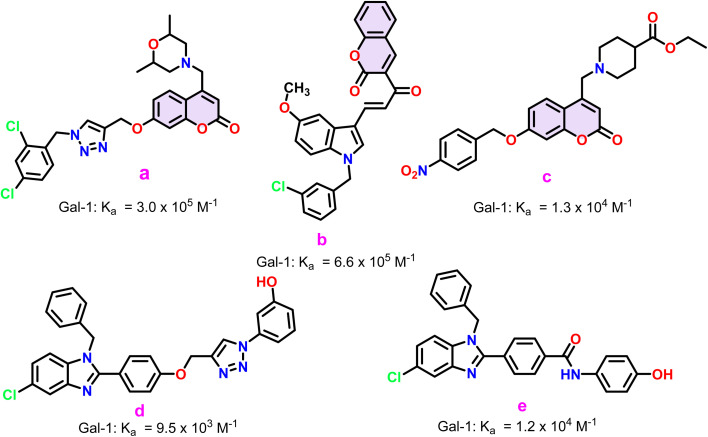
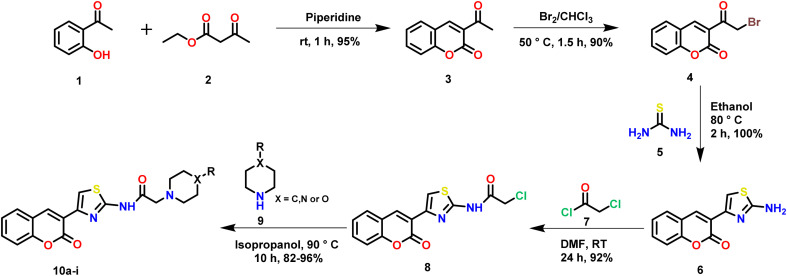
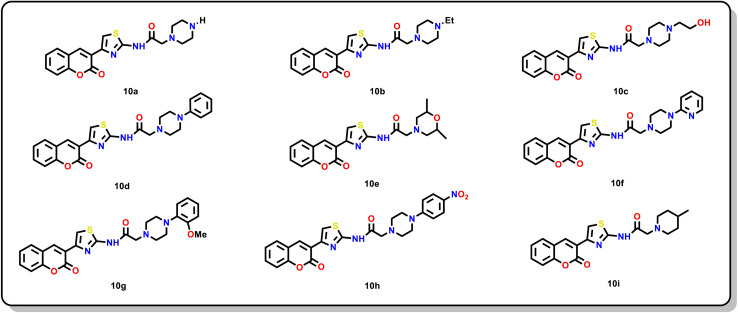
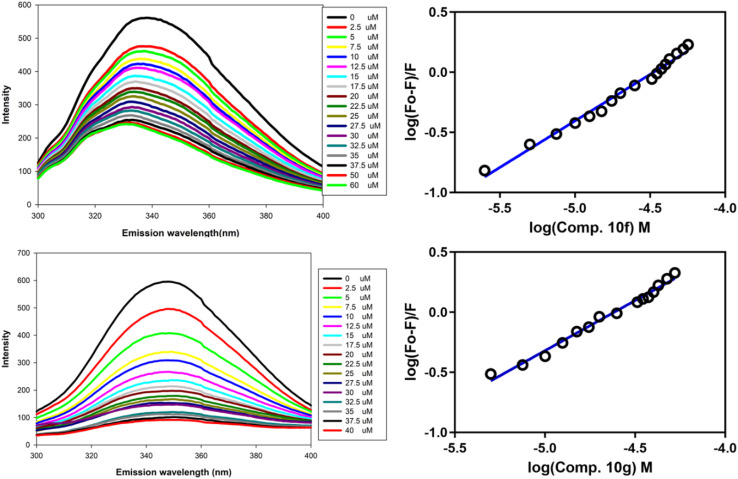
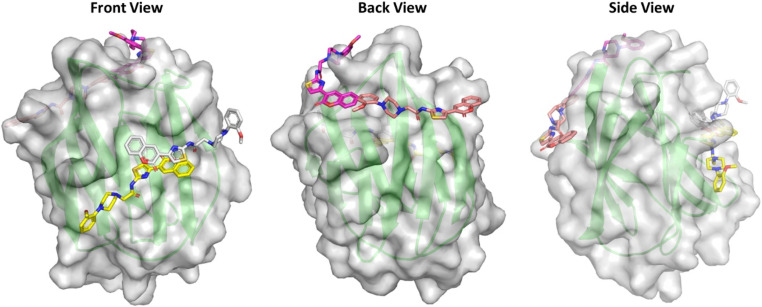
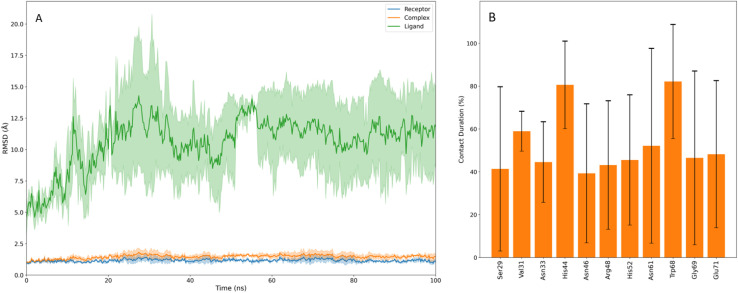
Sugar mimics are valuable tools in medicinal chemistry, offering the potential to overcome the limitations of carbohydrate inhibitors, such as poor pharmacokinetics and non-selectivity. In our continued efforts to develop heterocyclic galectin-1 inhibitors, we report the synthesis and characterization of thiazole-linked coumarin piperazine hybrids (10a-10i) as Gal-1 inhibitors. The compounds were characterized using H NMR, C NMR and HRMS. Among the synthesized molecules, four compounds demonstrated significant inhibitory activity, with more than 50% inhibition observed at a concentration of 20 μM in a Gal-1 enzyme assay. Fluorescence spectroscopy was further utilized to elucidate the binding constant for the synthesized compounds. 10g exhibited the highest affinity for Gal-1, with a binding constant ( ) of 9.8 × 10 M. To elucidate the mode of binding, we performed extensive computational analyses with 10g, including 1.2 μs all-atom molecular dynamics simulations coupled with a robust machine learning tool. Our findings indicate that 10g binds to the carbohydrate binding site of Gal-1, with the coumarin moiety playing a key role in binding interactions. Additionally, our study underscores the limitations of relying solely on docking scores for conformational selection and highlights the critical importance of performing multiple MD replicas to gain accurate insights.
糖类模拟物是药物化学中的重要工具,具有克服碳水化合物抑制剂局限性的潜力,如不良的药代动力学和非选择性。在我们持续开发杂环半乳糖凝集素-1抑制剂的工作中,我们报道了噻唑连接的香豆素哌嗪杂化物(10a - 10i)作为Gal - 1抑制剂的合成与表征。这些化合物通过氢核磁共振、碳核磁共振和高分辨质谱进行表征。在合成的分子中,四种化合物表现出显著的抑制活性,在Gal - 1酶测定中,浓度为20 μM时观察到超过50%的抑制率。进一步利用荧光光谱法阐明合成化合物的结合常数。10g对Gal - 1表现出最高亲和力,结合常数( )为9.8 × 10 M。为了阐明结合模式,我们对10g进行了广泛的计算分析,包括1.2微秒的全原子分子动力学模拟以及一个强大的机器学习工具。我们的研究结果表明,10g与Gal - 1的碳水化合物结合位点结合,香豆素部分在结合相互作用中起关键作用。此外,我们的研究强调了仅依靠对接分数进行构象选择的局限性,并突出了进行多个分子动力学复制品以获得准确见解的至关重要性。