Wang Chenghui, Zhang Liyao, Nie Zhipeng, Liang Min, Liu Hanqing, Yi Qiuzi, Wang Chunyan, Ai Cheng, Zhang Juanjuan, Gao Yinglong, Ji Yanchun, Guan Min-Xin
Center for Mitochondrial Biomedicine and Department of Ophthalmology, the Fourth Affiliated Hospital.
Department of Genetics, and.
JCI Insight. 2024 Nov 19;10(1):e182209. doi: 10.1172/jci.insight.182209.
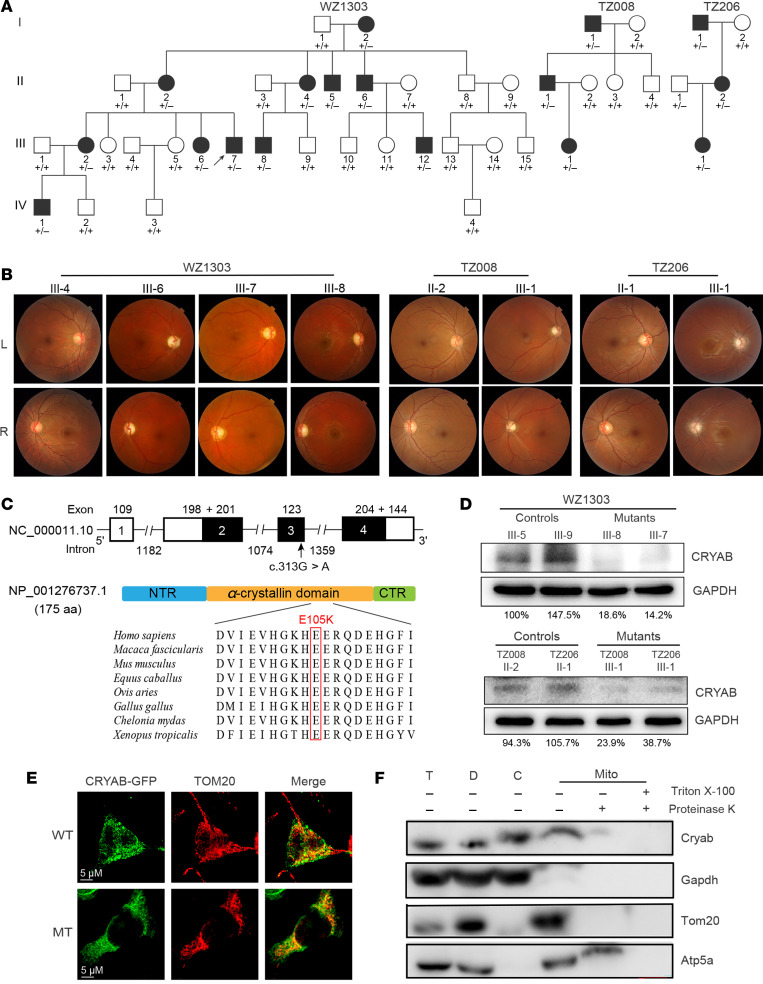
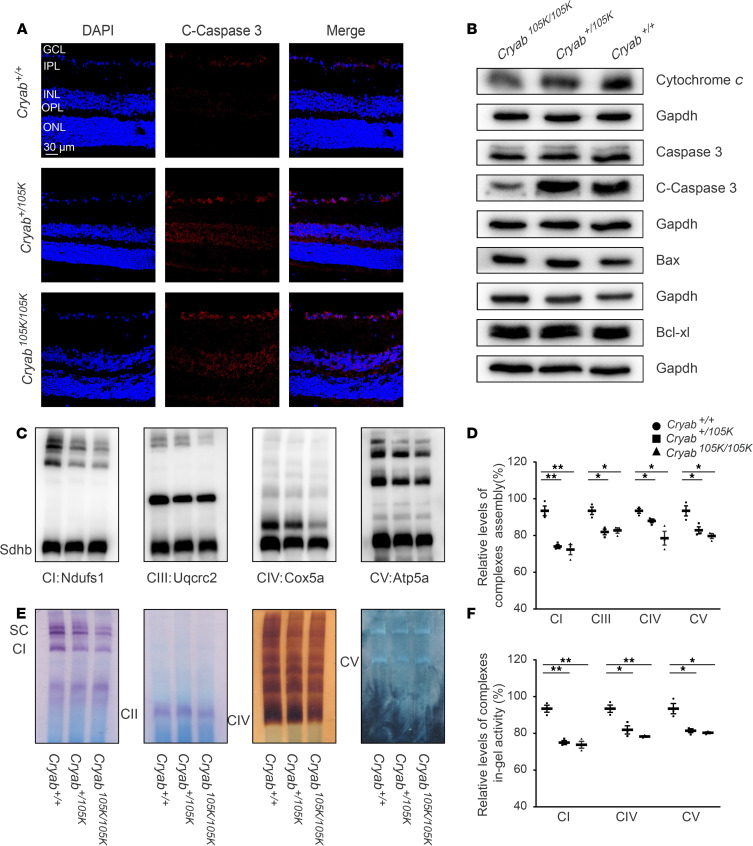
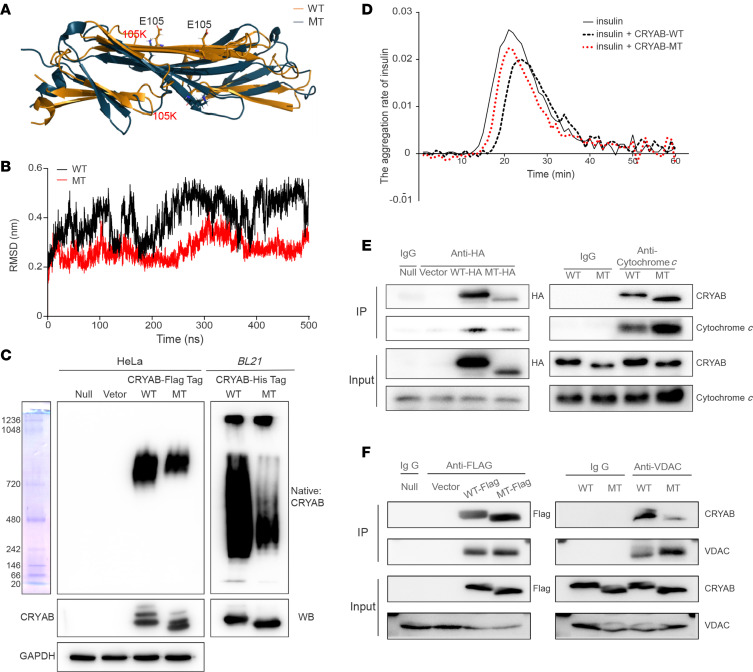
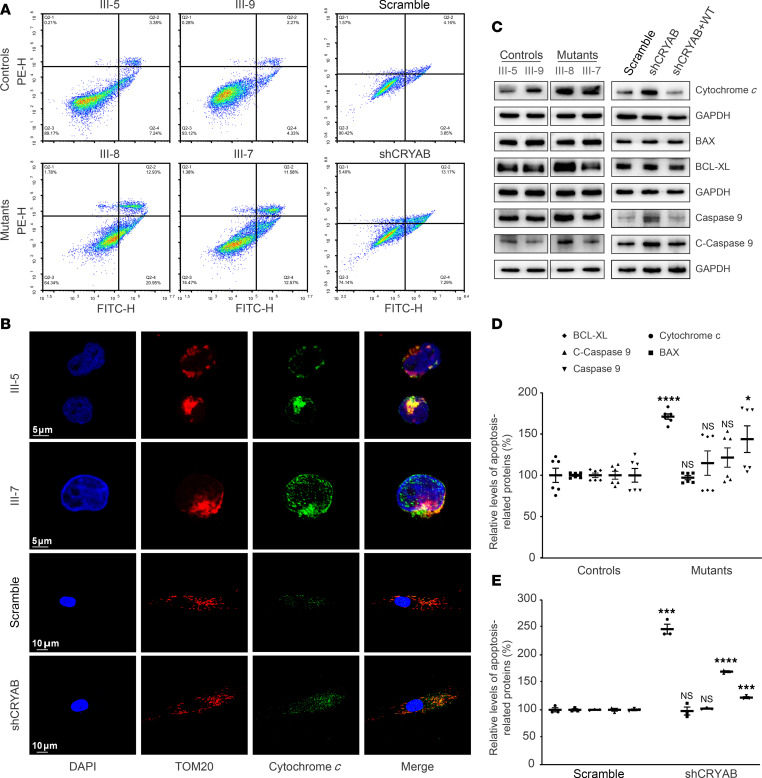
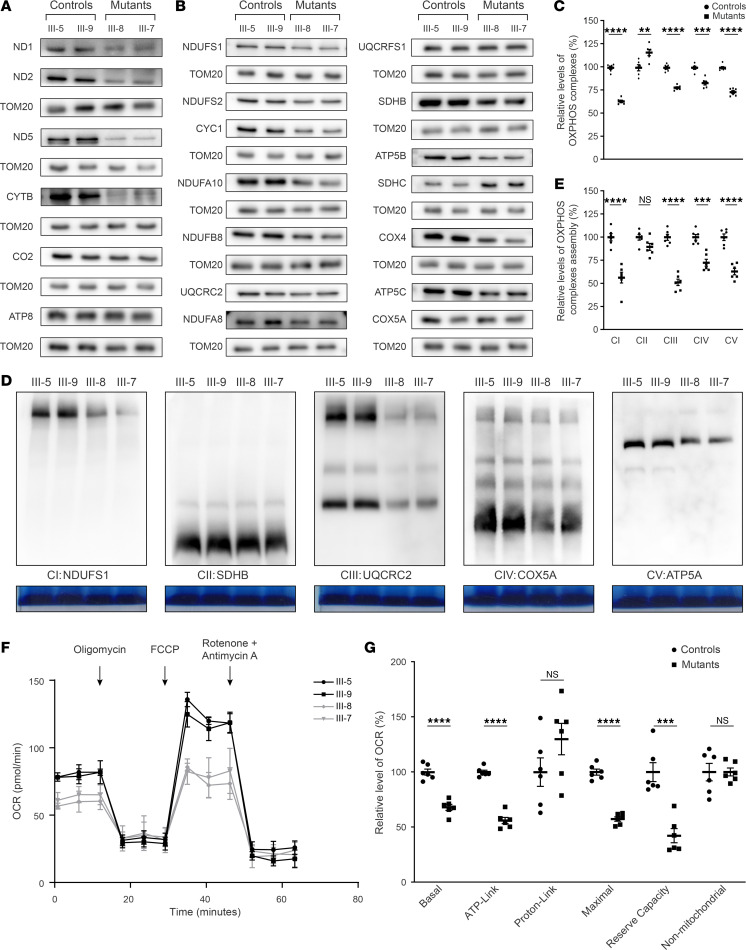
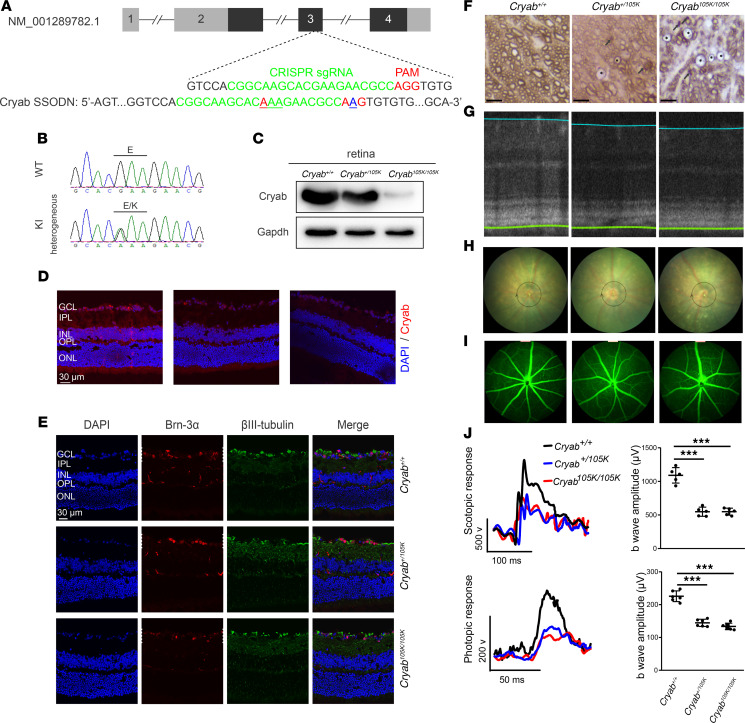
The degeneration of retinal ganglion cells (RGC) due to mitochondrial dysfunctions manifests optic neuropathy. However, the molecular components of RGC linked to optic neuropathy manifestations remain largely unknown. Here, we identified a potentially novel optic atrophy-causative CRYAB gene encoding a highly conserved major lens protein acting as mitochondrial chaperone and possessing antiapoptotic activities. The heterozygous CRYAB mutation (c.313G>A, p. Glu105Lys) was cosegregated with autosomal dominant inheritance of optic atrophy in 3 Chinese families. The p.E105K mutation altered the structure and function of CRYAB, including decreased stability, reduced formation of oligomers, and decreased chaperone activity. Coimmunoprecipitation indicated that the p.E105K mutation reduced the interaction of CRYAB with apoptosis-associated cytochrome c and voltage-dependent anion channel protein. The cell lines carrying the p.E105K mutation displayed promotion of apoptosis and defective assembly, stability, and activities of oxidative phosphorylation system as well as imbalance of mitochondrial dynamics. Involvement of CRYAB in optic atrophy was confirmed by phenotypic evaluations of Cryabp.E105K-knockin mice. These mutant mice exhibited ocular lesions that included alteration of intraretinal layers, degeneration of RGCs, photoreceptor deficits, and abnormal retinal vasculature. Furthermore, Cryab-deficient mice displayed elevated apoptosis and mitochondrial dysfunctions. Our findings provide insight of pathophysiology of optic atrophy arising from RGC degeneration caused by CRYAB deficiency-induced elevated apoptosis and mitochondrial dysfunctions.
视网膜神经节细胞(RGC)因线粒体功能障碍而退化会导致视神经病变。然而,与视神经病变表现相关的RGC分子成分在很大程度上仍然未知。在此,我们鉴定出一个潜在的新型致视神经萎缩的CRYAB基因,该基因编码一种高度保守的主要晶状体蛋白,作为线粒体伴侣并具有抗凋亡活性。杂合CRYAB突变(c.313G>A,p.Glu105Lys)在3个中国家系中与视神经萎缩的常染色体显性遗传共分离。p.E105K突变改变了CRYAB的结构和功能,包括稳定性降低、寡聚体形成减少以及伴侣活性降低。免疫共沉淀表明,p.E105K突变减少了CRYAB与凋亡相关细胞色素c和电压依赖性阴离子通道蛋白的相互作用。携带p.E105K突变的细胞系表现出凋亡增加以及氧化磷酸化系统的组装、稳定性和活性缺陷,还有线粒体动力学失衡。通过对Cryabp.E105K敲入小鼠的表型评估证实了CRYAB与视神经萎缩有关。这些突变小鼠表现出眼部病变,包括视网膜内层改变、RGC退化、光感受器缺陷以及视网膜血管异常。此外,Cryab基因缺陷小鼠表现出凋亡增加和线粒体功能障碍。我们的研究结果为因CRYAB缺陷导致的凋亡增加和线粒体功能障碍引起的RGC退化所致视神经萎缩的病理生理学提供了见解。