Rizwan Khadija, Galbraith John Morrison
Department of Chemistry, Biochemistry and Physics, Marist College, 3399 North Road, Poughkeepsie, NY 12601, USA.
Molecules. 2024 Nov 15;29(22):5396. doi: 10.3390/molecules29225396.
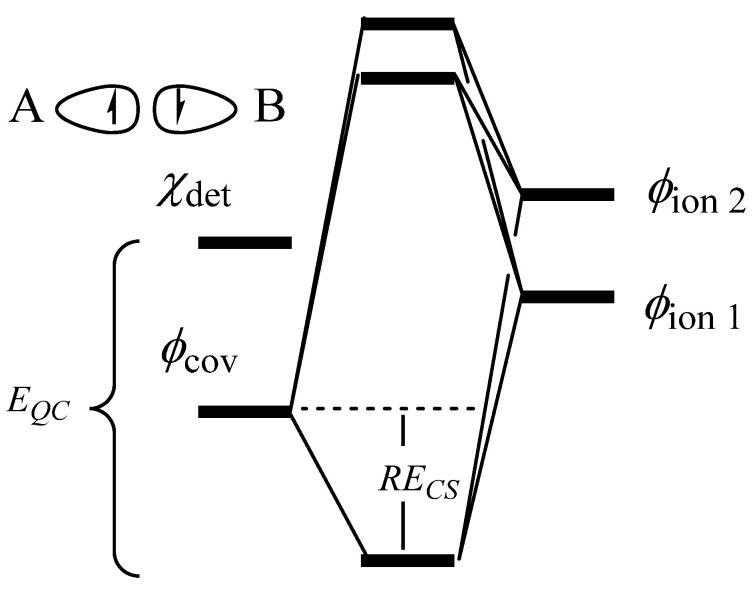
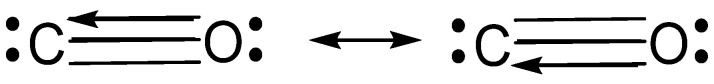
Valence bond theory (VB) was used to determine the extent and driving forces for covalent vs. dative bonding in 10-valence-electron diatomic molecules N, CO, NO, CN, P, SiS, PS, and SiP. VBSCF calculations were performed at the CCSD(T)/cc-pVDZ optimized geometries. The full triply bonded system included 20 VB structures. A separation of the σ and π space allowed for a subdivision of the full 20 structure set into sets of 8 and 3 for the π and σ systems, respectively. The smaller structure sets allowed for a more focused look at each type of bond. In situ bond energies for σ bonds, individual π bonds, the π system, and triple bonds follow expected trends. Our data shows that N and P have three covalent bonds whereas CO and SiS contain two covalent and one dative bond, and charged species NO, CN, PS, and SiP are a mixture of N and CO type electronic arrangements, resulting in a nearly equal charge distribution. Dative bonds prefer to be in the π position due to enhanced σ covalency and π resonance. Both σ and π resonance energies depend on a balance of ionic strength, orbital compactness, σ constraints, and bond directionality. Resonance energy is a major contributor to bond strength, making up more than 50% of the π bonds in SiS and PS (charge-shift bonds), and is greater than charge transfer in dative bonds.
价键理论(VB)用于确定具有10个价电子的双原子分子N、CO、NO、CN、P、SiS、PS和SiP中,共价键与配位键的程度及驱动力。VBSCF计算在CCSD(T)/cc-pVDZ优化几何结构下进行。完整的三键体系包含20个VB结构。将σ和π空间分离后,可将完整的20个结构集分别细分为π体系的8个结构集和σ体系的3个结构集。较小的结构集有助于更专注地研究每种键型。σ键、单个π键、π体系和三键的原位键能遵循预期趋势。我们的数据表明,N和P有三个共价键,而CO和SiS包含两个共价键和一个配位键,带电物种NO、CN、PS和SiP是N型和CO型电子排布的混合物,导致电荷分布几乎相等。由于σ共价性增强和π共振,配位键更倾向于处于π位置。σ和π共振能均取决于离子强度、轨道紧凑性、σ限制和键的方向性之间的平衡。共振能是键强度的主要贡献因素,在SiS和PS(电荷转移键)的π键中占比超过50%,且大于配位键中的电荷转移。