Mignani Renzo, Biagini Elena, Cianci Vittoria, Pieruzzi Federico, Pisani Antonio, Tuttolomondo Antonino, Pieroni Maurizio
Nephrology, Dialysis and Transplantation, IRCCS S. Orsola Hospital Bologna, University of Bologna, Bologna, Italy.
Cardiology Unit, Cardiac Thoracic and Vascular Department, IRCCS Azienda Ospedaliero-Universitaria di Bologna, Bologna, Italy.
Adv Ther. 2025 Feb;42(2):597-635. doi: 10.1007/s12325-024-03041-2. Epub 2024 Dec 5.
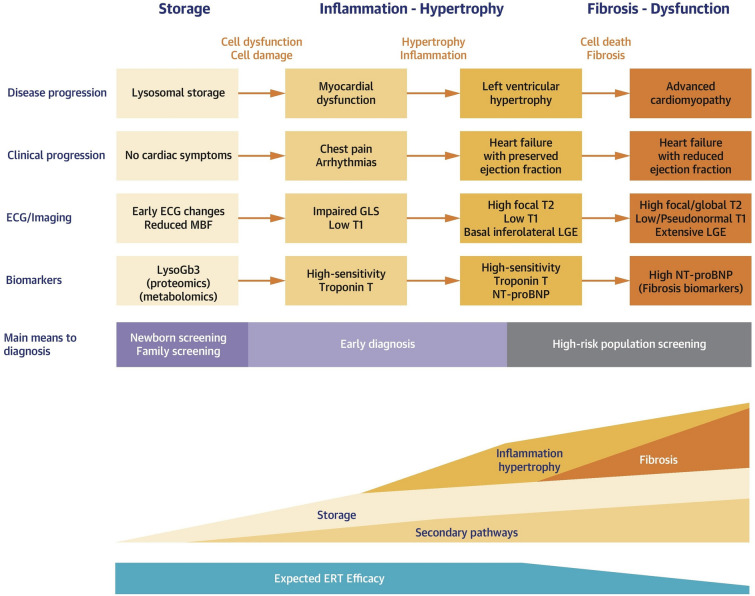
Fabry disease (FD) is a rare lysosomal storage disorder that is characterized by renal, neurological, and cardiovascular dysfunction. Four treatments are currently available for patients with FD; three enzyme replacement therapies (ERTs; agalsidase alfa, agalsidase beta, and pegunigalsidase alfa) and one pharmacological chaperone (migalastat). This review focuses on the evidence for the benefits of ERTs and migalastat, and provides an overview of their impact on disease manifestations and quality of life (QoL). Agalsidase beta is associated with renal, neurological, and cardiovascular benefits, and may prevent renal disease progression. Agalsidase alfa provides stabilizing effects across all main organ systems, although minor sex-specific differences exist in patients with more advanced baseline disease. The benefits of agalsidase alfa and agalsidase beta are similar but depend on the extent of baseline disease. Some data indicate that agalsidase beta may be preferable over the longer term. Both agalsidase alfa and agalsidase beta are associated with improved gastrointestinal and pain symptoms, as well as improved QoL. Patients with advanced end-organ damage tend not to respond as optimally to ERTs as those who initiate ERTs before irreversible organ fibrosis develops, highlighting the need for early treatment initiation. Migalastat, which is only approved for patients with amenable missense gene variants, generally stabilizes renal parameters and provides cardiovascular benefits. Migalastat also improves diarrhea and pain, and stabilizes QoL (although ERT may be more effective for pain management), but the neurological effects of migalastat have not been studied. Real-world data raise concerns about effective in vivo amenability of some genetic variants. Future studies with direct treatment comparisons in patients with FD are needed.
法布里病(FD)是一种罕见的溶酶体贮积症,其特征为肾脏、神经和心血管功能障碍。目前有四种治疗方法可供FD患者使用;三种酶替代疗法(ERTs;阿加糖酶α、阿加糖酶β和聚乙二醇化阿加糖酶α)和一种药理伴侣(米加司他)。本综述重点关注ERTs和米加司他的益处证据,并概述它们对疾病表现和生活质量(QoL)的影响。阿加糖酶β对肾脏、神经和心血管有益,可能会阻止肾脏疾病进展。阿加糖酶α对所有主要器官系统都有稳定作用,尽管在基线疾病较严重的患者中存在轻微的性别差异。阿加糖酶α和阿加糖酶β的益处相似,但取决于基线疾病的程度。一些数据表明,从长期来看,阿加糖酶β可能更具优势。阿加糖酶α和阿加糖酶β都与胃肠道和疼痛症状改善以及QoL提高相关。终末器官严重受损的患者对ERTs的反应往往不如在不可逆器官纤维化发生之前开始ERTs治疗的患者理想,这凸显了早期开始治疗的必要性。米加司他仅被批准用于有合适错义基因变异的患者,通常可稳定肾脏参数并带来心血管益处。米加司他还可改善腹泻和疼痛,并稳定QoL(尽管ERT在疼痛管理方面可能更有效),但尚未研究米加司他的神经学作用。真实世界数据引发了对某些基因变异体内有效适应性的担忧。需要对FD患者进行直接治疗比较的未来研究。