Pironon Nathalie, Gasparyan Artyom, Yubero María Joao, Duchatelet Sabine, Hovhannesyan Kristina, Leclerc-Mercier Stephanie, Kostandyan Natella, Palisson Francis, Sarkisian Tamara, Titeux Matthias, Fuentes Ignacia, Hovnanian Alain
Laboratory of Genetic Skin Diseases, Institut Imagine, Université Paris Cité, Inserm, UMR 1163, F-75015, Paris, France.
Center of Medical Genetics and Primary Health Care, Abovyan Street, Yerevan, Armenia.
Eur J Hum Genet. 2025 Mar;33(3):344-350. doi: 10.1038/s41431-024-01717-5. Epub 2024 Dec 5.
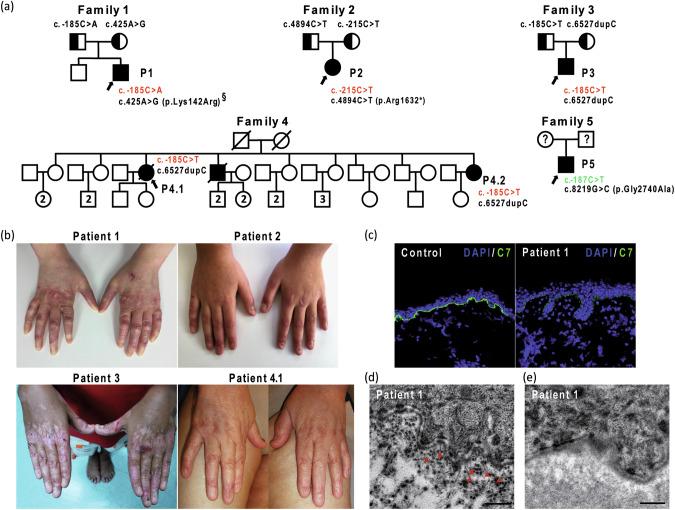
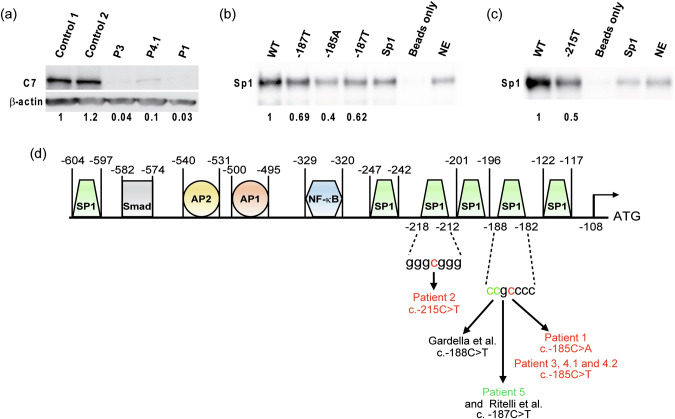
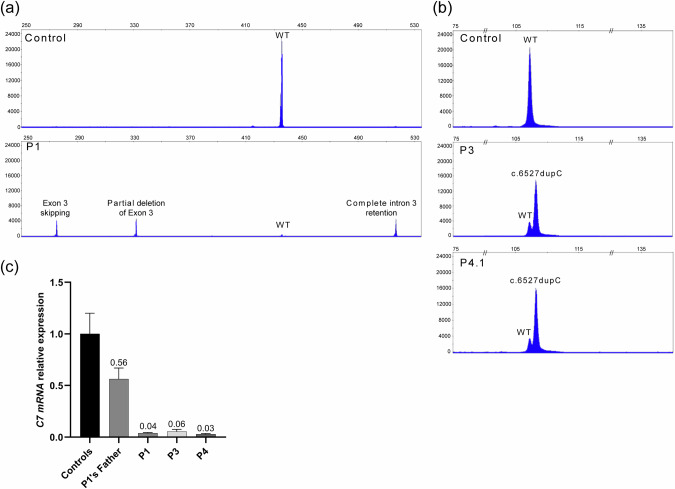
Recessive dystrophic epidermolysis bullosa (RDEB) is a rare and most often severe genodermatosis characterized by recurrent blistering and erosions of the skin and mucous membranes after minor trauma, leading to major local and systemic complications. RDEB is caused by loss-of-function mutations in COL7A1 encoding type VII collagen (C7), the main component of anchoring fibrils which form attachment structures stabilizing the cutaneous basement membrane zone. Most of the previously reported COL7A1 mutations are located in the coding or intronic regions. We describe 6 patients with localized or intermediate RDEB for whom one recessive pathogenic variant in the coding region and a second variant in the COL7A1 promoter were identified. These substitutions, three of which are novel, are localized in two Sp1 binding sites of the promoter region. DNA pull-down assay showed a drastic reduction of Sp1 binding consistent with a dramatic decrease in COL7A1 transcript and almost undetectable C7 protein levels. Our results reveal that mutations in the COL7A1 promoter on the background of a null allele can underlie localized or intermediate RDEB. They further emphasize the functional importance of Sp1 motifs in the proximal COL7A1 promoter which should be carefully investigated for regulatory mutations in the case of RDEB with only one pathogenic variant identified in the coding or intronic regions.
隐性营养不良性大疱性表皮松解症(RDEB)是一种罕见且通常较为严重的遗传性皮肤病,其特征为轻微创伤后皮肤和黏膜反复出现水疱和糜烂,进而导致严重的局部和全身并发症。RDEB由编码VII型胶原蛋白(C7)的COL7A1功能丧失性突变引起,C7是构成锚定纤维的主要成分,而锚定纤维形成的附着结构可稳定皮肤基底膜带。先前报道的大多数COL7A1突变位于编码区或内含子区域。我们描述了6例局限性或中间型RDEB患者,在这些患者中,一个编码区隐性致病变异和COL7A1启动子中的第二个变异被鉴定出来。这些替代突变中有三个是新发现的,它们位于启动子区域的两个Sp1结合位点。DNA下拉实验显示Sp1结合显著减少,这与COL7A1转录本急剧下降以及几乎检测不到的C7蛋白水平一致。我们的结果表明,在无效等位基因背景下COL7A1启动子中的突变可能是局限性或中间型RDEB的病因。这些结果进一步强调了Sp1基序在COL7A1近端启动子中的功能重要性,对于在编码区或内含子区域仅鉴定出一个致病变异的RDEB病例,应仔细研究其调控突变。