Orozco-Arias Simon, Sierra Pío, Durbin Richard, González Josefa
Institute of Evolutionary Biology, CSIC, UPF, 08003 Barcelona, Spain.
Department of Genetics, University of Cambridge, Cambridge CB2 3EH, United Kingdom.
Genome Res. 2024 Dec 23;34(12):2256-2268. doi: 10.1101/gr.278821.123.
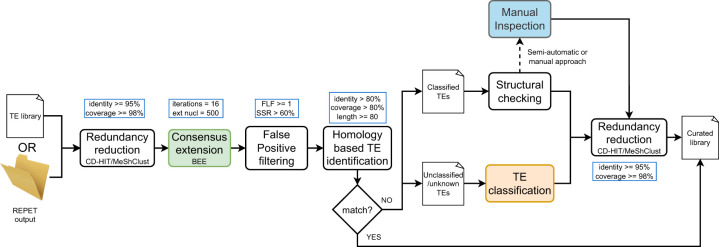
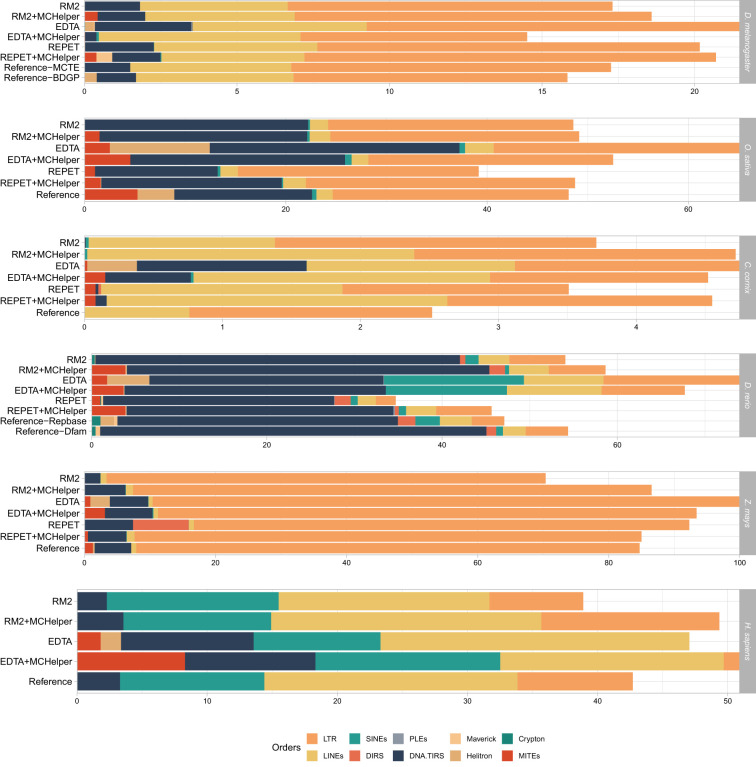
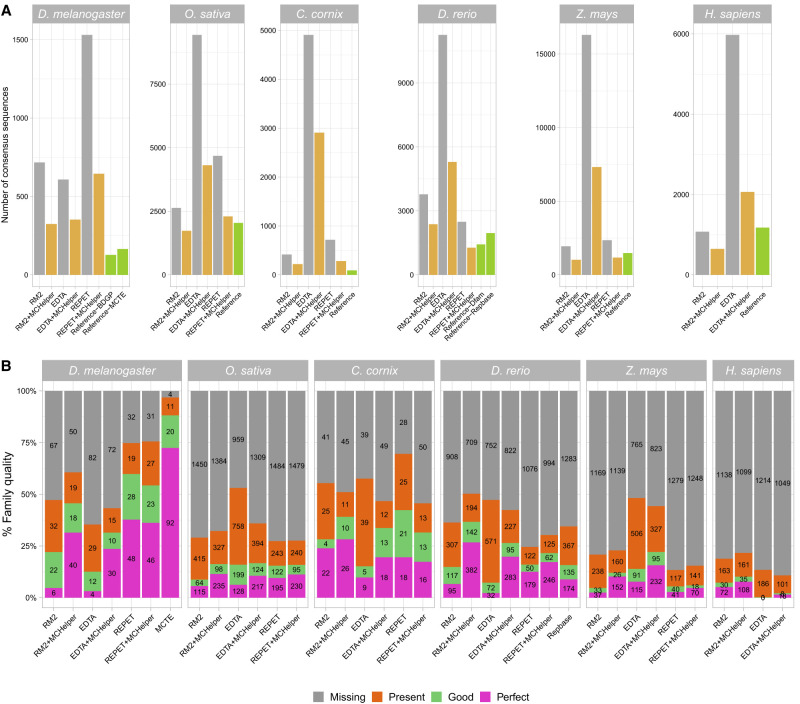
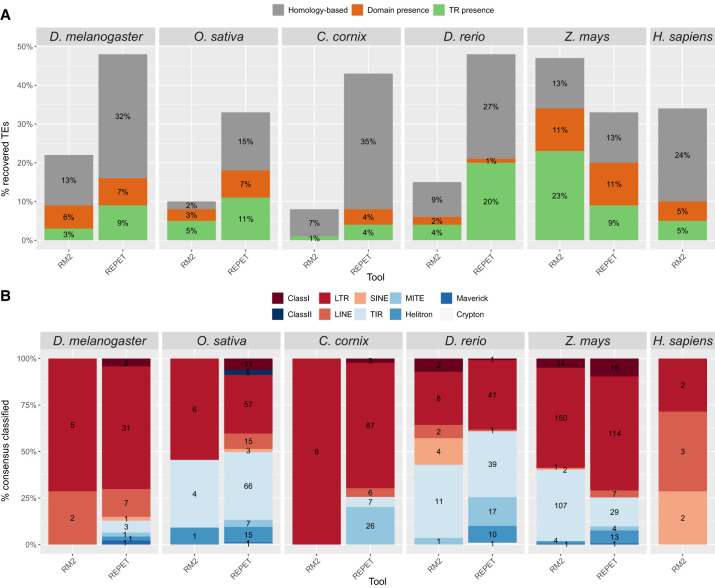
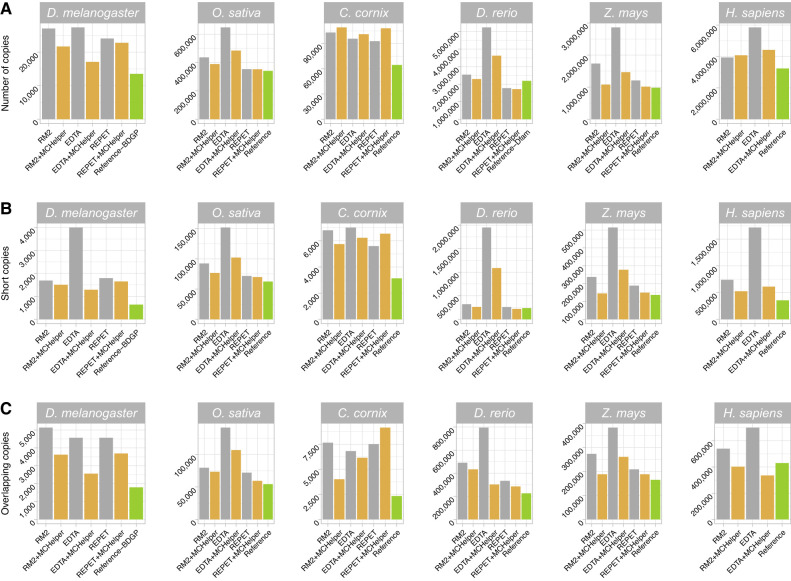
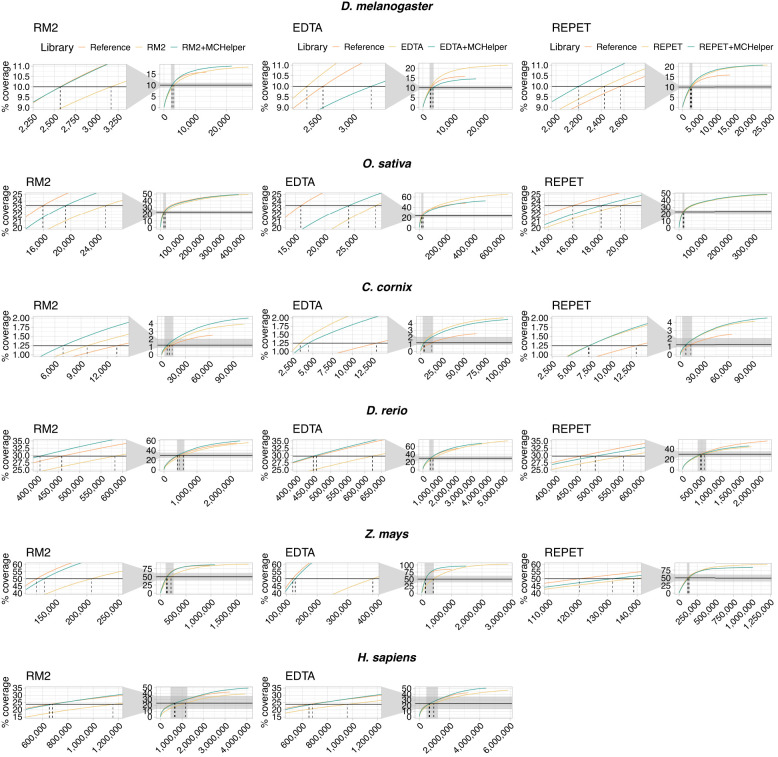
The number of species with high-quality genome sequences continues to increase, in part due to the scaling up of multiple large-scale biodiversity sequencing projects. While the need to annotate genic sequences in these genomes is widely acknowledged, the parallel need to annotate transposable element (TE) sequences that have been shown to alter genome architecture, rewire gene regulatory networks, and contribute to the evolution of host traits is becoming ever more evident. However, accurate genome-wide annotation of TE sequences is still technically challenging. Several de novo TE identification tools are now available, but manual curation of the libraries produced by these tools is needed to generate high-quality genome annotations. Manual curation is time-consuming, and thus impractical for large-scale genomic studies, and lacks reproducibility. In this work, we present the Manual Curator Helper tool MCHelper, which automates the TE library curation process. By leveraging MCHelper's fully automated mode with the outputs from three de novo TE identification tools, RepeatModeler2, EDTA, and REPET, in the fruit fly, rice, hooded crow, zebrafish, maize, and human, we show a substantial improvement in the quality of the TE libraries and genome annotations. MCHelper libraries are less redundant, with up to 65% reduction in the number of consensus sequences, have up to 11.4% fewer false positive sequences, and up to ∼48% fewer "unclassified/unknown" TE consensus sequences. Genome-wide TE annotations are also improved, including larger unfragmented insertions. Moreover, MCHelper is an easy-to-install and easy-to-use tool.
拥有高质量基因组序列的物种数量持续增加,部分原因是多个大规模生物多样性测序项目的规模扩大。虽然人们普遍认识到需要对这些基因组中的基因序列进行注释,但对转座元件(TE)序列进行注释的同等需求也日益明显,因为转座元件已被证明会改变基因组结构、重塑基因调控网络并推动宿主性状的进化。然而,在全基因组范围内准确注释TE序列在技术上仍然具有挑战性。现在有几种从头识别TE的工具,但需要对这些工具生成的文库进行人工整理,以生成高质量的基因组注释。人工整理耗时,因此对于大规模基因组研究不切实际,而且缺乏可重复性。在这项工作中,我们展示了人工整理辅助工具MCHelper,它能自动执行TE文库整理过程。通过在果蝇、水稻、 hooded crow、斑马鱼、玉米和人类中利用MCHelper的全自动模式以及三种从头识别TE工具RepeatModeler2、EDTA和REPET的输出结果,我们发现TE文库和基因组注释的质量有了显著提高。MCHelper文库的冗余度更低,共有序列数量减少了65%,假阳性序列减少了11.4%,“未分类/未知”的TE共有序列减少了约48%。全基因组TE注释也得到了改善,包括更大的未断裂插入。此外,MCHelper是一个易于安装和使用的工具。