Martinez Fernandez Jose, Haji Seyed Javadi Alireza, Teat Simon J, Cundari Thomas R, Tilley T Don
Department of Chemistry, University of California, Berkeley, Berkeley, California 94720, United States.
Department of Chemistry, Center for Advanced Scientific Computing and Modeling (CASCaM), University of North Texas, Denton, Texas 76203, United States.
J Am Chem Soc. 2024 Dec 18;146(50):34962-34969. doi: 10.1021/jacs.4c14642. Epub 2024 Dec 10.
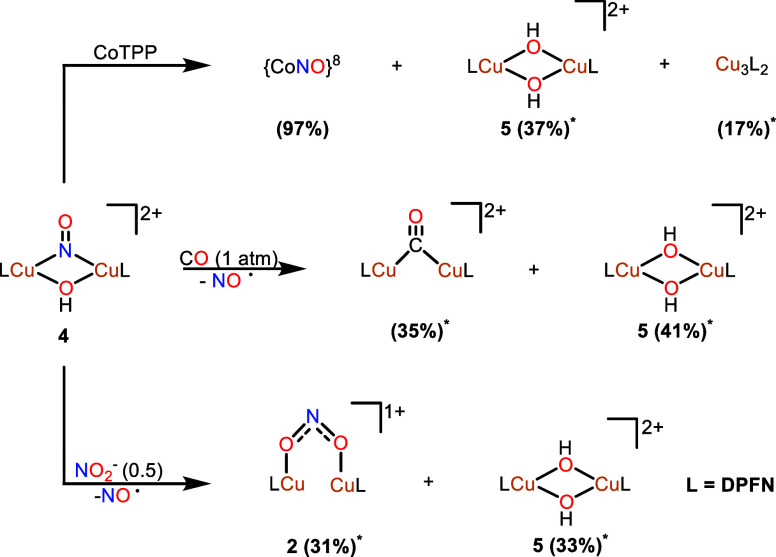
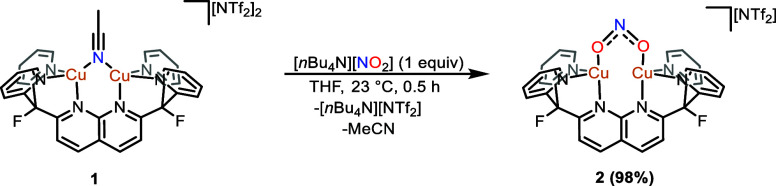
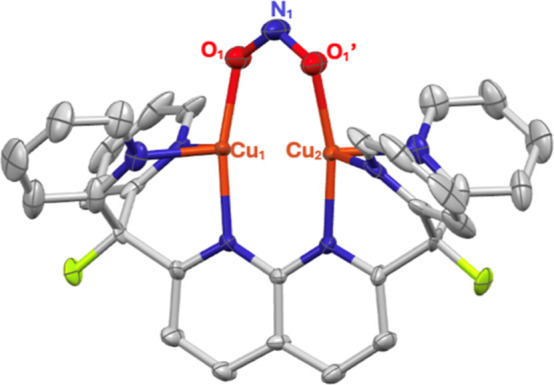
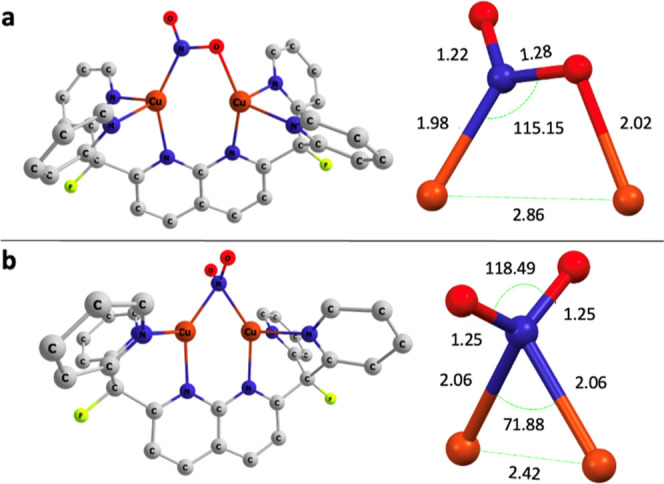
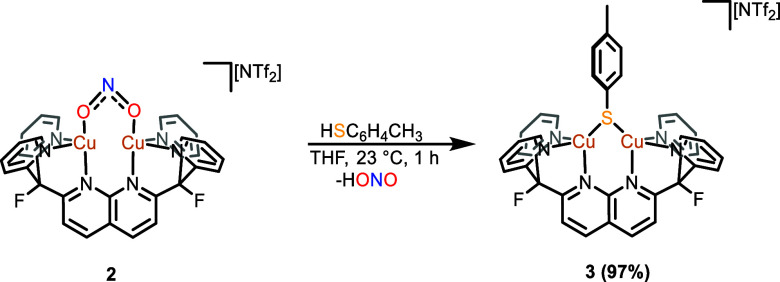
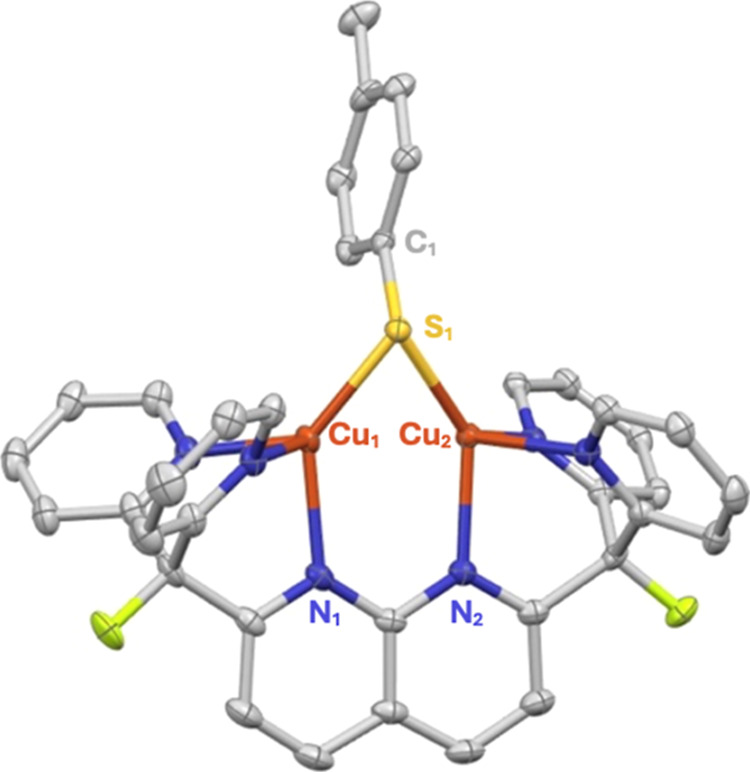
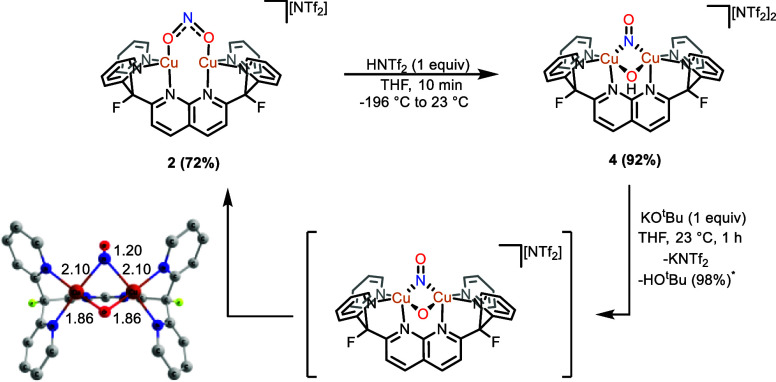
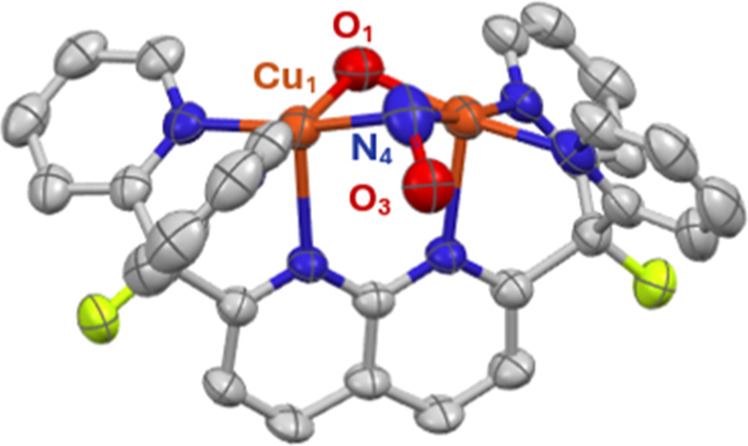
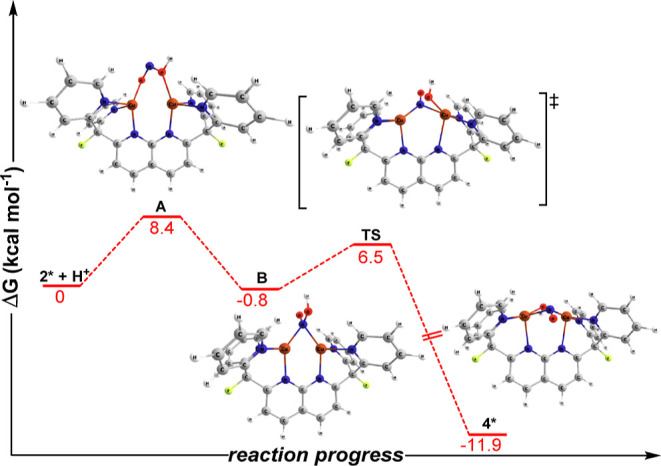
A monocationic dicopper(I,I) nitrite complex [Cu(μ-κ:κ-ON)DPFN][NTf] () (DPFN = 2,7-bis(fluoro-di(2-pyridyl)methyl)-1,8-naphthyridine, NTf = N(SOCF)), was synthesized by treatment of a dicopper acetonitrile complex, [Cu(μ-MeCN)DPFN][NTf] (), with tetrabutylammonium nitrite ([BuN][NO]). DFT calculations indicate that is one of three linkage isomers that are close in energy and presumably accessible in solution. Reaction of the μ-κ:κ-ON complex with -TolSH produces nitrous acid (HONO) and the corresponding dicopper thiolate species via an acid-base exchange reaction. Notably, treatment of with HNTf results in N-O bond cleavage in the putative, HONO-ligated complex to form the more thermodynamically favorable nitrosyl-bridged dicopper complex [Cu(μ-NO)(μ-OH)DPFN][NTf] (). This scission can be reversed via deprotonation of the hydroxy ligand with KOBu. X-ray diffraction studies confirmed the solid-state molecular structures of and . DFT calculations were used to construct a reaction coordinate diagram detailing formation of the μ-NO complex and to describe its electronic structure. The nitrosyl ligand in is chemically labile, as demonstrated by its ready displacement in reactions with CO or NO.
通过用亚硝酸四丁铵([Bu₄N][NO₂])处理二铜乙腈配合物[Cu(μ-MeCN)DPFN][NTf₂](),合成了一种单阳离子二铜(I,I)亚硝酸盐配合物[Cu(μ-κ:κ-ONO)DPFN][NTf₂]()(DPFN = 2,7-双(氟-二(2-吡啶基)甲基)-1,8-萘啶,NTf₂ = N(SO₂CF₃)₂)。密度泛函理论计算表明,是能量相近且可能在溶液中可获得的三种键合异构体之一。μ-κ:κ-ONO配合物与对甲苯硫醇(p-TolSH)反应通过酸碱交换反应生成亚硝酸(HONO)和相应的二铜硫醇盐物种。值得注意的是,用HNTf₂处理会导致假定的HONO配位配合物中的N-O键断裂,形成热力学上更有利的亚硝酰基桥联二铜配合物[Cu(μ-NO)(μ-OH)DPFN][NTf₂]()。通过用叔丁醇钾(KOtBu)使羟基配体去质子化,这种断裂可以逆转。X射线衍射研究证实了和的固态分子结构。密度泛函理论计算用于构建详细描述μ-NO配合物形成的反应坐标图,并描述其电子结构。中的亚硝酰基配体在化学上不稳定,这在与CO或NO的反应中很容易被取代就证明了这一点。