Sugawara Ryota, Ito Hidenori, Tabata Hidenori, Ueda Hiroshi, Scala Marcello, Nagata Koh-Ichi
Department of Molecular Neurobiology, Institute for Developmental Research, Aichi Developmental Disability Center, 713-8 Kamiya, Kasugai 480-0392, Japan.
United Graduate School of Drug Discovery and Medical Information Sciences, Gifu University, Yanagido 1-1, Gifu 501-1193, Japan.
Cells. 2024 Dec 9;13(23):2032. doi: 10.3390/cells13232032.
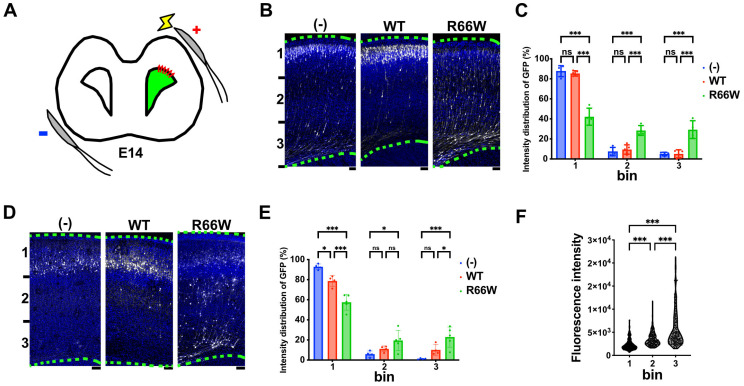
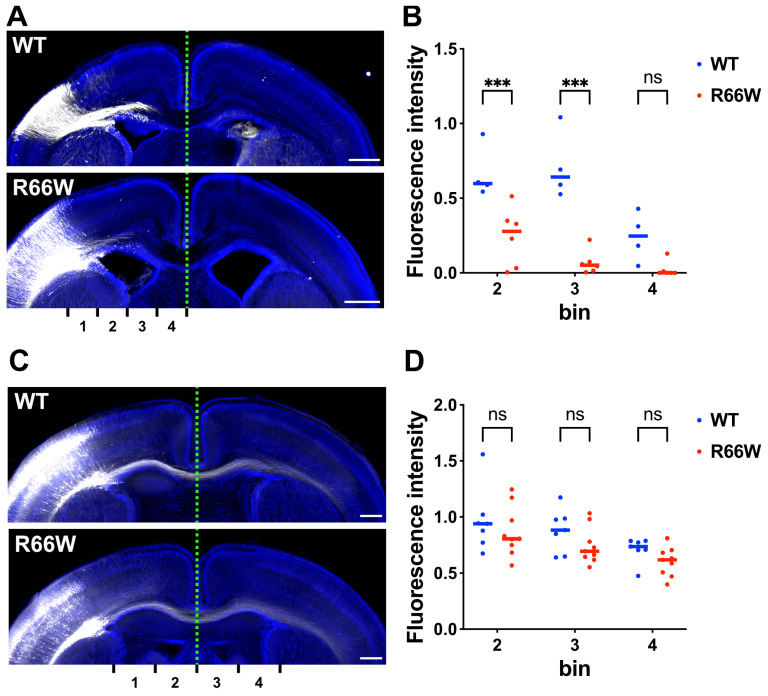
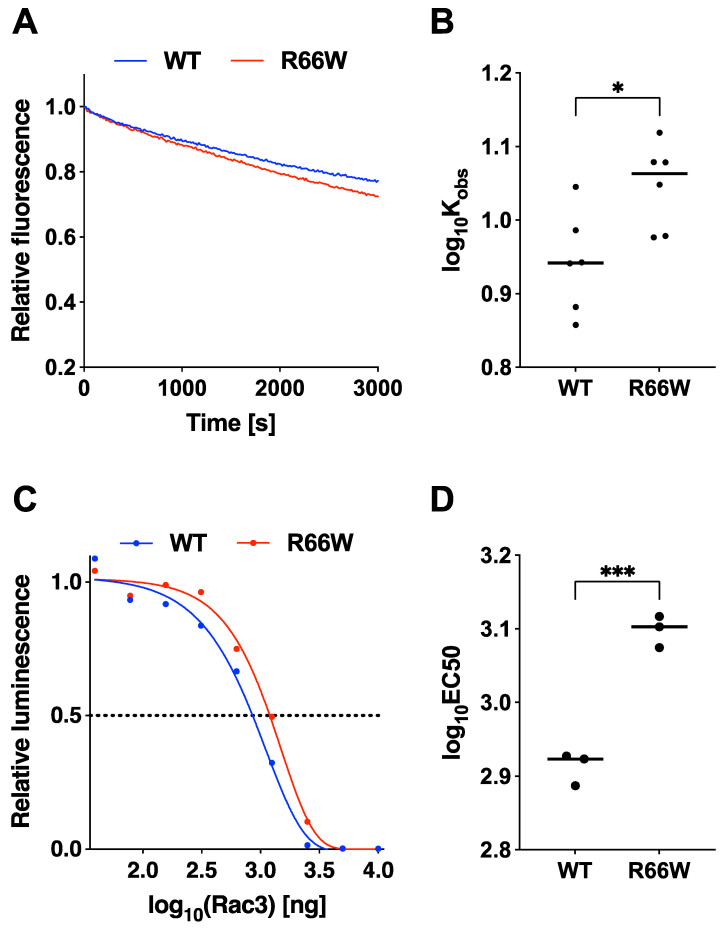
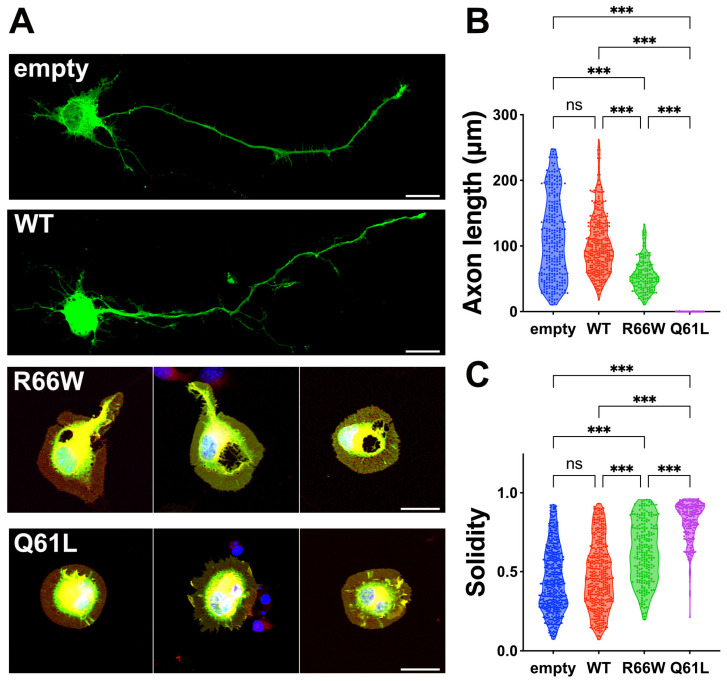
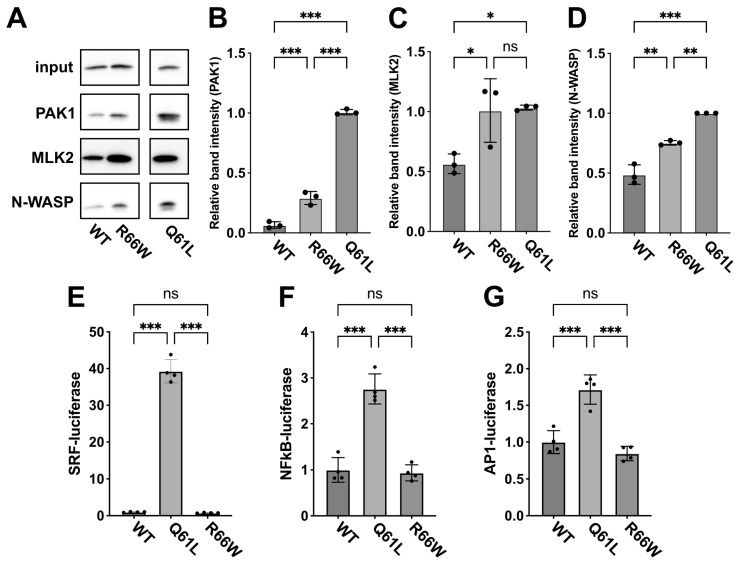
encodes a small GTPase of the Rho family that plays a critical role in actin cytoskeleton remodeling and intracellular signaling regulation. Pathogenic variants in , all of which reported thus far affect conserved residues within its functional domains, have been linked to neurodevelopmental disorders characterized by diverse phenotypic features, including structural brain anomalies and facial dysmorphism (NEDBAF). Recently, a novel de novo variant (NM_005052.3): c.196C>T, p.R66W was identified in a prenatal case with fetal akinesia deformation sequence (a spectrum of conditions that interfere with the fetus's ability to move), and complex brain malformations featuring corpus callosum agenesis, diencephalosynapsis, kinked brainstem, and vermian hypoplasia. To investigate the mechanisms underlying the association between RAC3 deficiency and this unique, distinct clinical phenotype, we explored the pathophysiological significance of the p.R66W variant in brain development. Biochemical assays revealed a modest enhancement in intrinsic GDP/GTP exchange activity and an inhibitory effect on GTP hydrolysis. Transient expression studies in COS7 cells demonstrated that RAC3-R66W interacts with the downstream effectors PAK1, MLK2, and N-WASP but fails to activate SRF-, AP1-, and NFkB-mediated transcription. Additionally, overexpression of RAC3-R66W significantly impaired differentiation in primary cultured hippocampal neurons. Acute expression of RAC3-R66W in vivo by in utero electroporation resulted in impairments in cortical neuron migration and axonal elongation during corticogenesis. Collectively, these findings suggest that the p.R66W variant may function as an activated version in specific signaling pathways, leading to a distinctive and severe prenatal phenotype through variant-specific mechanisms.
编码一种Rho家族的小GTPase,它在肌动蛋白细胞骨架重塑和细胞内信号调节中起关键作用。该基因的致病变异,迄今为止报道的所有变异均影响其功能域内的保守残基,已与以多种表型特征为特征的神经发育障碍相关联,包括脑结构异常和面部畸形(NEDBAF)。最近,在一例产前病例中发现了一种新的从头变异(NM_005052.3):c.196C>T,p.R66W,该病例患有胎儿运动不能变形序列(一系列干扰胎儿运动能力的病症),以及具有胼胝体发育不全、间脑联合、脑干扭结和小脑蚓部发育不全的复杂脑畸形。为了研究RAC3缺乏与这种独特、明显的临床表型之间关联的潜在机制,我们探讨了p.R66W变异在脑发育中的病理生理意义。生化分析显示内在GDP/GTP交换活性有适度增强,对GTP水解有抑制作用。在COS7细胞中的瞬时表达研究表明,RAC3-R66W与下游效应器PAK1、MLK2和N-WASP相互作用,但未能激活SRF-、AP1-和NFkB介导的转录。此外,RAC3-R66W的过表达显著损害原代培养海马神经元的分化。通过子宫内电穿孔在体内急性表达RAC3-R66W导致皮质发生过程中皮质神经元迁移和轴突伸长受损。总的来说,这些发现表明p.R66W变异可能在特定信号通路中作为激活形式发挥作用,通过变异特异性机制导致独特且严重的产前表型。