Naru Jasmine, Othus Megan, Lin ChenWei, Biernacki Melinda A, Bleakley Marie, Chauncey Thomas R, Erba Harry P, Fang Min, Fitzgibbon Matthew P, Gafken Phillip R, Ivey Richard G, Kennedy Jacob J, Lorentzen Travis D, Meshinchi Soheil, Moseley Anna, Pogosova-Agadjanyan Era L, Liu Vivian M, Radich Jerald P, Voytovich Uliana J, Wang Pei, Whiteaker Jeffrey R, Willman Cheryl L, Wu Feinan, Paulovich Amanda G, Stirewalt Derek L
Translational Science and Therapeutics Division Fred Hutch Seattle Washington USA.
SWOG Statistics and Data Management Center Fred Hutch Seattle Washington USA.
EJHaem. 2024 Oct 25;5(6):1243-1251. doi: 10.1002/jha2.1041. eCollection 2024 Dec.
Acute myeloid leukemia (AML) remains one of the deadliest hematopoietic malignancies. A better understanding of the molecular biology governing AML may lead to improved risk stratification and facilitate the development of novel therapies. Proteins are responsible for much of the biology of cells. Several studies have examined the global proteome in bulk mononuclear cells (MNCs) from AML specimens, which are comprised a heterogenous population of cells at various stages of differentiation.
Given the potential impact of the nonleukemic cells on protein expression profiles, we applied an integrative proteogenomic approach utilizing next-generation sequencing and mass spectrometry-based proteomics to identify novel protein biomarkers in unsorted MNCs and viable leukemic blasts (VLBs) isolated from blood and bone marrow specimens obtained at the time of AML diagnosis.
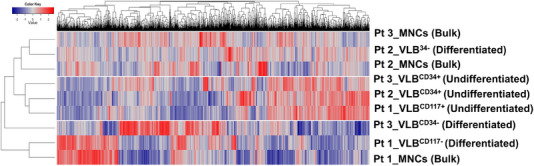
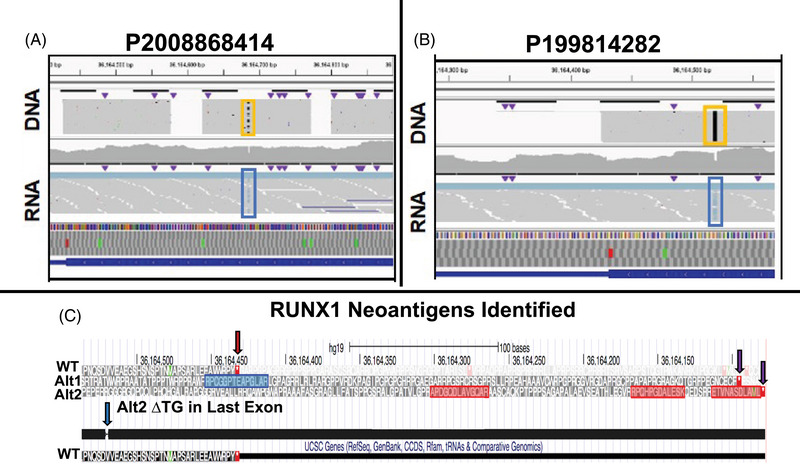
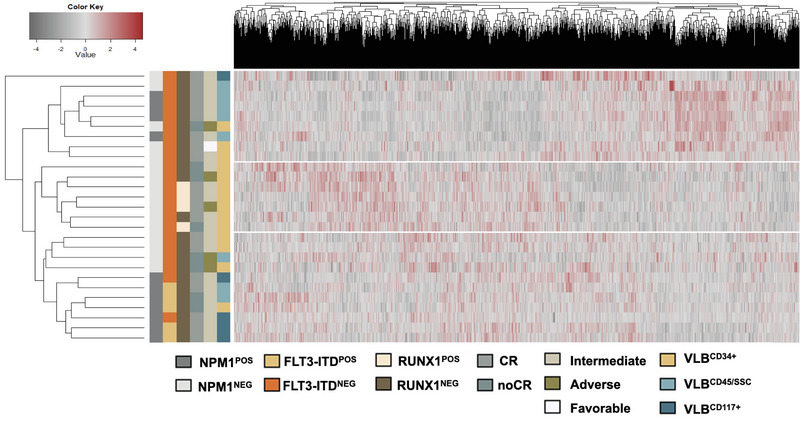
We identified significant differences in protein expression between VLBs and MNCs. Subsequent studies ( = 27) focused on proteomic profiling of VLBs that identified novel candidate biomarkers associated with mutational genotypes and clinical outcome, some of which were recapitulated in an independent cohort of patients. Using mass spectrometry, we also detected mutated protein products, some of which were predicted via in silico analyses to be potential neoantigens amenable to adoptive immunotherapy. As previously described, analyses comparing transcript and protein expression showed an overall modest correlation between mRNA and protein dataset, but enriching for genes associated with mutations significantly improved the protein-RNA correlation.
Together, the results provide insight into the biology of VLBs and demonstrate the gains derived from examining the proteome in addition to genome and transcriptome.
急性髓系白血病(AML)仍然是最致命的造血系统恶性肿瘤之一。更好地理解AML的分子生物学机制可能会改善风险分层,并促进新型疗法的开发。蛋白质在细胞的许多生物学过程中发挥着作用。多项研究已对AML标本中的大量单核细胞(MNC)进行了全蛋白质组分析,这些细胞是处于不同分化阶段的异质细胞群体。
鉴于非白血病细胞对蛋白质表达谱可能产生的影响,我们采用了一种整合蛋白质基因组学方法,利用下一代测序和基于质谱的蛋白质组学技术,在未分选的MNC以及从AML诊断时获取的血液和骨髓标本中分离出的存活白血病原始细胞(VLB)中鉴定新型蛋白质生物标志物。
我们发现VLB和MNC之间存在蛋白质表达的显著差异。后续研究(n = 27)聚焦于VLB的蛋白质组分析,确定了与突变基因型和临床结果相关的新型候选生物标志物,其中一些在独立患者队列中得到了验证。通过质谱分析,我们还检测到了突变的蛋白质产物,其中一些经计算机分析预测为适合过继性免疫治疗的潜在新抗原。如前所述,比较转录本和蛋白质表达的分析表明,mRNA和蛋白质数据集之间总体呈适度相关性,但富集与突变相关的基因显著改善了蛋白质 - RNA相关性。
总之,这些结果为VLB的生物学特性提供了见解,并证明了除基因组和转录组外,研究蛋白质组所带来的收获。