Glaser Selina, Kretzmer Helene, Kolassa Iris Tatjana, Schlesner Matthias, Fischer Anja, Fenske Isabell, Siebert Reiner, Ammerpohl Ole
Institute of Human Genetics, Ulm University and Ulm University Medical Center, Albert-Einstein-Allee 11, Ulm 89081, Germany.
Department of Genome Regulation, Max Planck Institute for Molecular Genetics, Ihnestraße 63-73, Berlin 14195, Germany.
NAR Genom Bioinform. 2024 Dec 18;6(4):lqae181. doi: 10.1093/nargab/lqae181. eCollection 2024 Dec.
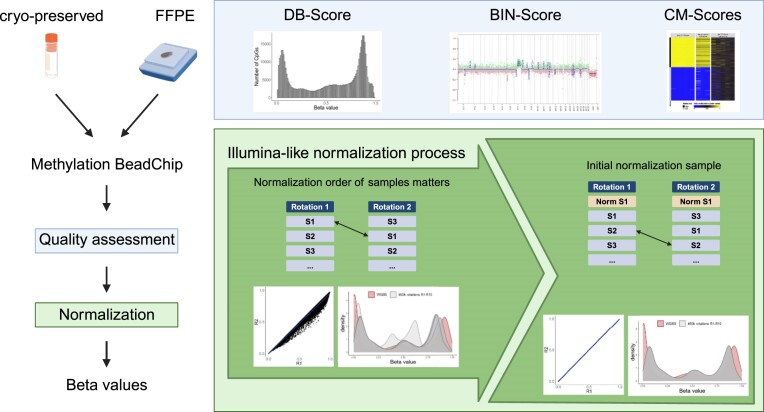
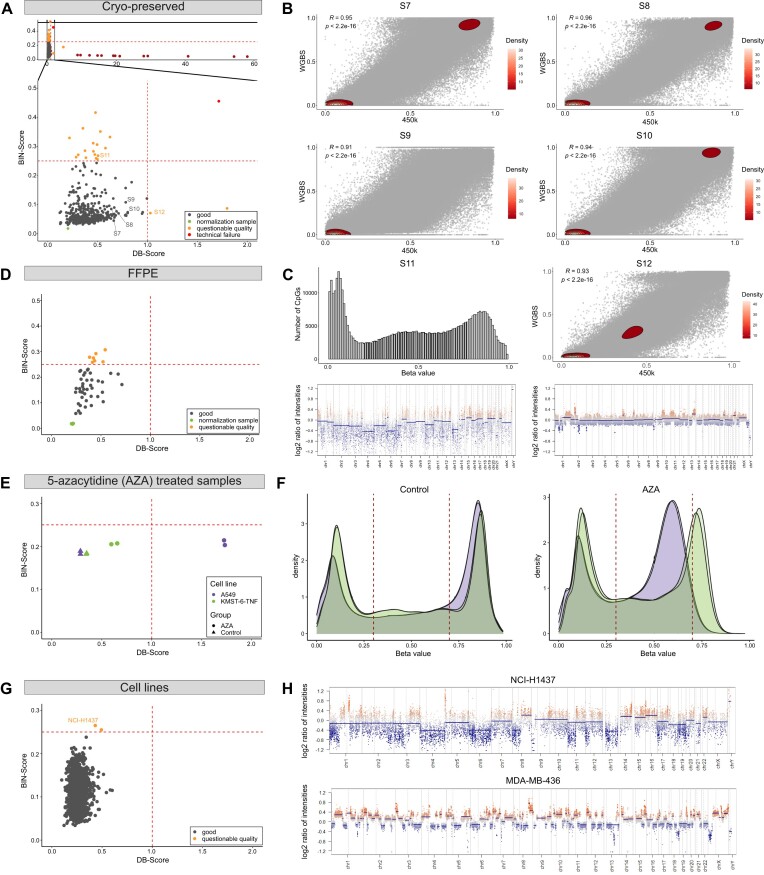
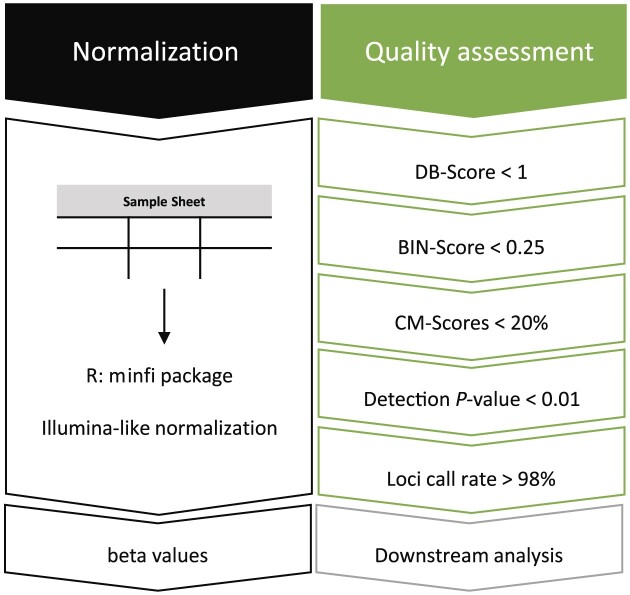
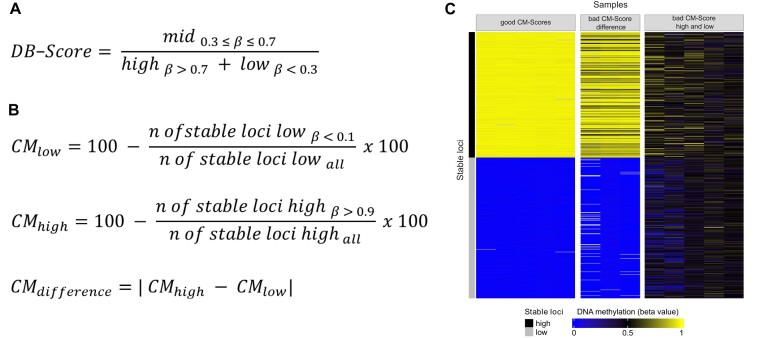
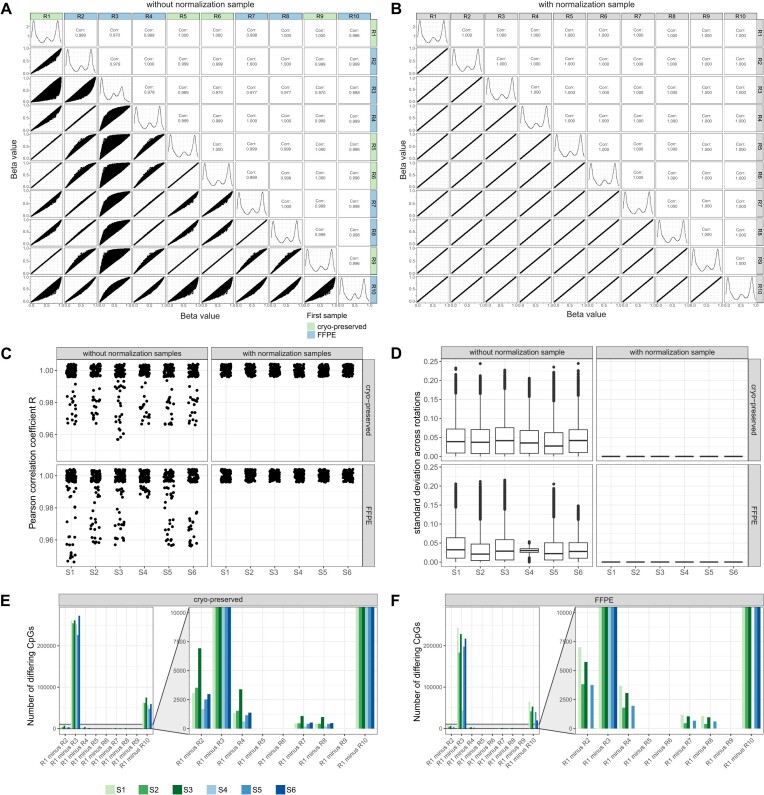
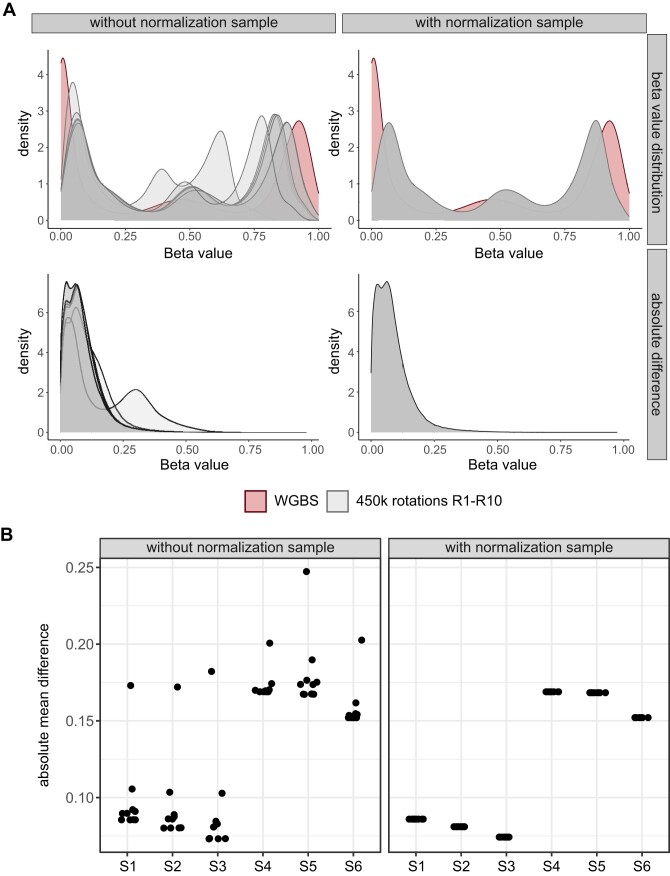
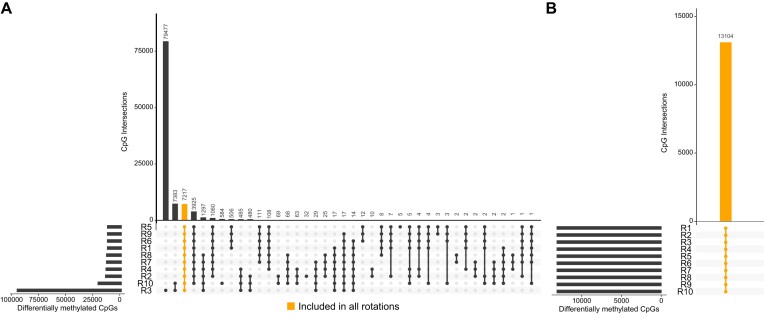
Illumina-based BeadChip arrays have revolutionized genome-wide DNA methylation profiling, pushing it into diagnostics. However, comprehensive quality assessment remains challenging within a wide range of available tissue materials and sample preparation methods. This study tackles two critical issues: differentiating between biological effects and technical artefacts in suboptimal quality samples and the impact of the first sample on the Illumina-like normalization algorithm. We introduce three quality control scores based on global DNA methylation distribution (DB-Score), bin distance from copy number variation analysis (BIN-Score) and consistently methylated CpGs (CM-Score) that rely on biological features rather than internal array controls. These scores, designed to be adjustable for different analysis tools and sample cohort characteristics, were explored and benchmarked across independent cohorts. Additionally, we reveal deviations in beta values caused by different sample rankings with the Illumina-like normalization algorithm, verified these with whole-genome methylation sequencing data and showed effects on differential DNA methylation analysis. Our findings underscore the necessity of consistently utilizing a pre-defined normalization sample within the ranking process to boost reproducibility of the Illumina-like normalization algorithm. Overall, our study delivers valuable insights, practical recommendations and R functions designed to enhance reproducibility and quality assurance of DNA methylation analysis, particularly for challenging sample types.
基于Illumina的BeadChip阵列彻底改变了全基因组DNA甲基化谱分析,将其推向了诊断领域。然而,在广泛的可用组织材料和样本制备方法中,全面的质量评估仍然具有挑战性。本研究解决了两个关键问题:区分质量欠佳样本中的生物学效应和技术假象,以及第一个样本对类似Illumina归一化算法的影响。我们引入了基于全局DNA甲基化分布的三个质量控制分数(DB分数)、来自拷贝数变异分析的bin距离(BIN分数)和持续甲基化的CpG(CM分数),这些分数依赖于生物学特征而非阵列内部对照。这些分数旨在针对不同的分析工具和样本队列特征进行调整,并在独立队列中进行了探索和基准测试。此外,我们揭示了类似Illumina归一化算法因不同样本排名导致的β值偏差,并用全基因组甲基化测序数据进行了验证,并展示了其对差异DNA甲基化分析的影响。我们的研究结果强调了在排名过程中始终使用预定义归一化样本以提高类似Illumina归一化算法可重复性的必要性。总体而言,我们的研究提供了有价值的见解、实用建议和R函数,旨在提高DNA甲基化分析的可重复性和质量保证,特别是对于具有挑战性的样本类型。