Olatoke Tasnim, Zhang Erik Y, Wagner Andrew, He Quan, Li Siru, Astreinidis Aristotelis, McCormack Francis X, Xu Yan, Yu Jane J
Department of Pharmacology, Physiology and Neurobiology, University of Cincinnati; Cincinnati, OH 45267, USA.
Division of Pulmonary, Critical Care, and Sleep Medicine, Department of Internal Medicine, University of Cincinnati; Cincinnati, OH 45267, USA.
bioRxiv. 2024 Dec 12:2024.12.11.627871. doi: 10.1101/2024.12.11.627871.
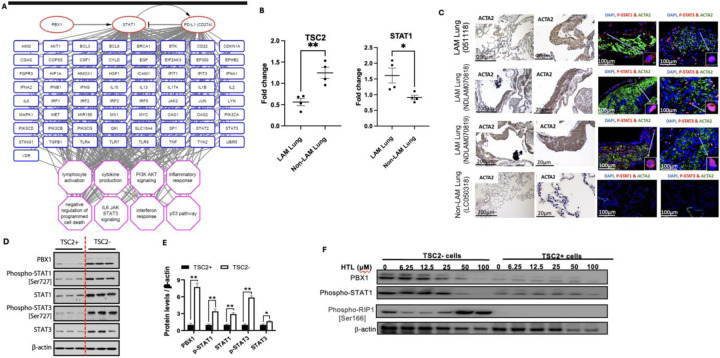
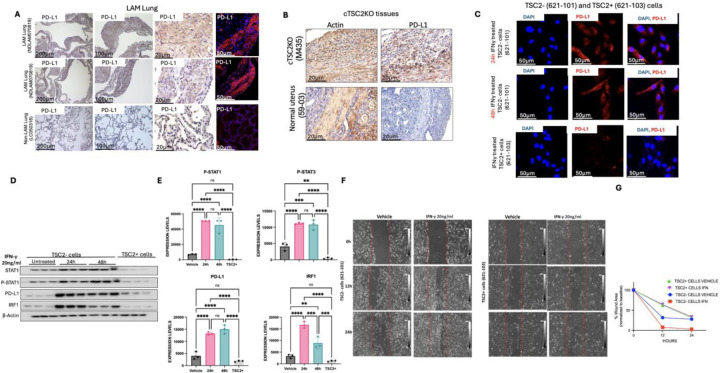
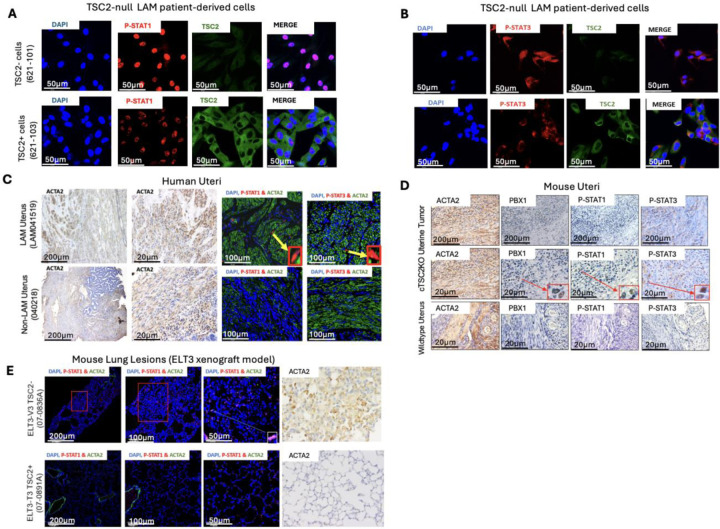
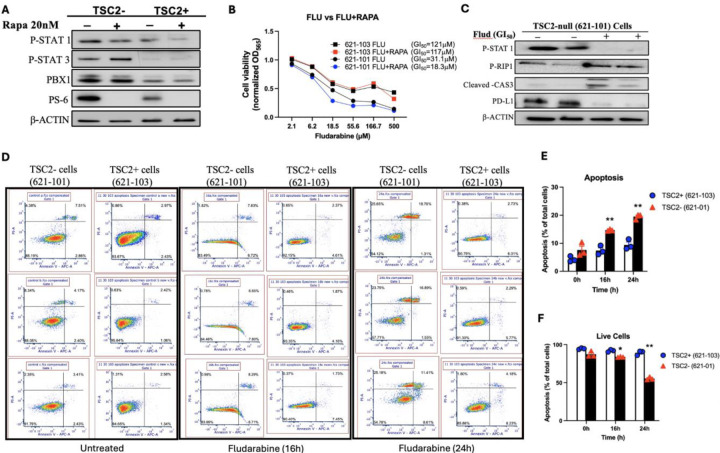
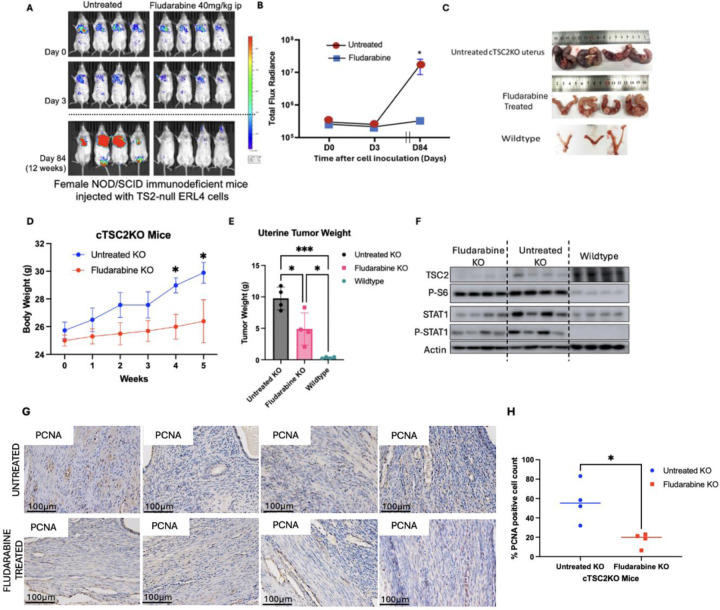
Lymphangioleiomyomatosis (LAM) is a cystic lung disease that primarily affects women. LAM is caused by the invasion of metastatic smooth muscle-like cells into the lung parenchyma, leading to abnormal cell proliferation, lung remodeling and progressive respiratory failure. LAM cells have TSC gene mutations, which occur sporadically or in people with Tuberous Sclerosis Complex. Although it is known that hyperactivation of the mechanistic target of rapamycin complex 1 (mTORC1) due to TSC2 gene mutations contributes to aberrant cell growth in LAM lung, tumor origin and invasive mechanism remain unclear. To determine molecular drivers responsible for aberrant LAM cell growth, we performed integrative single-cell transcriptomic analysis and predicted that STAT1 interacts with Pre-B cell leukemia transcription factor (PBX1) to regulate LAM cell survival. Here, we show activation of STAT1 and STAT3 proteins in TSC2-deficient LAM models. Fludarabine, a potent STAT1 inhibitor, induced the death of TSC2-deficient cells, increased caspase-3 cleavage, and phosphorylation of necroptosis marker RIP1. Fludarabine treatment impeded lung colonization of TSC2-deficient cells and uterine tumor progression, associated with reduced percentage of PCNA-positive cells in vivo. Interestingly, IFN-γ treatment increased STAT1 phosphorylation and PD-L1 expression, indicating that STAT1 aids TSC2-deficient tumor cells in evading immune surveillance in LAM. Our findings indicate that STAT1 signaling is critical for LAM cell survival and could be targeted to treat LAM and other mTORC1 hyperactive tumors.
淋巴管平滑肌瘤病(LAM)是一种主要影响女性的囊性肺部疾病。LAM是由转移性平滑肌样细胞侵入肺实质引起的,导致异常细胞增殖、肺重塑和进行性呼吸衰竭。LAM细胞存在TSC基因突变,这些突变可散发出现或发生在患有结节性硬化症复合体的人群中。尽管已知由于TSC2基因突变导致的雷帕霉素复合物1(mTORC1)机制性靶点的过度激活促成了LAM肺中的异常细胞生长,但肿瘤起源和侵袭机制仍不清楚。为了确定导致LAM细胞异常生长的分子驱动因素,我们进行了综合单细胞转录组分析,并预测信号转导和转录激活因子1(STAT1)与前B细胞白血病转录因子(PBX1)相互作用以调节LAM细胞存活。在此,我们展示了在TSC2缺陷的LAM模型中STAT1和信号转导和转录激活因子3(STAT3)蛋白的激活。氟达拉滨是一种有效的STAT1抑制剂,可诱导TSC2缺陷细胞死亡,增加半胱天冬酶-3的切割以及坏死性凋亡标志物受体相互作用蛋白1(RIP1)的磷酸化。氟达拉滨治疗阻碍了TSC2缺陷细胞在肺部的定植和子宫肿瘤进展,这与体内增殖细胞核抗原(PCNA)阳性细胞百分比降低有关。有趣的是,γ干扰素(IFN-γ)治疗增加了STAT1磷酸化和程序性死亡受体配体1(PD-L1)表达,表明STAT1有助于TSC2缺陷肿瘤细胞逃避免疫监视在LAM中的作用。我们的研究结果表明,STAT1信号传导对LAM细胞存活至关重要,并且可以作为治疗LAM和其他mTORC1过度活跃肿瘤的靶点。