Shahin-Shamsabadi Alireza, Cappuccitti John
Evolved.Bio, 280 Joseph Street, Kitchener, Ontario, Canada.
Heliyon. 2024 Nov 29;10(24):e40772. doi: 10.1016/j.heliyon.2024.e40772. eCollection 2024 Dec 30.
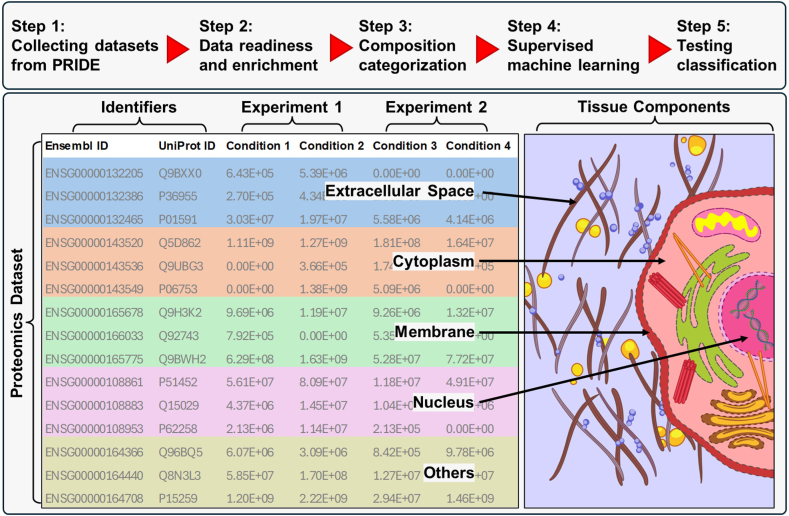
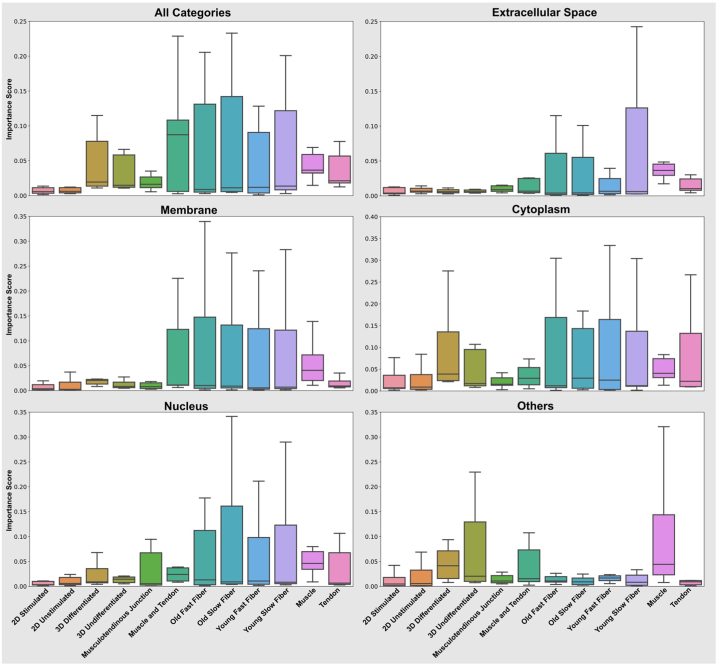
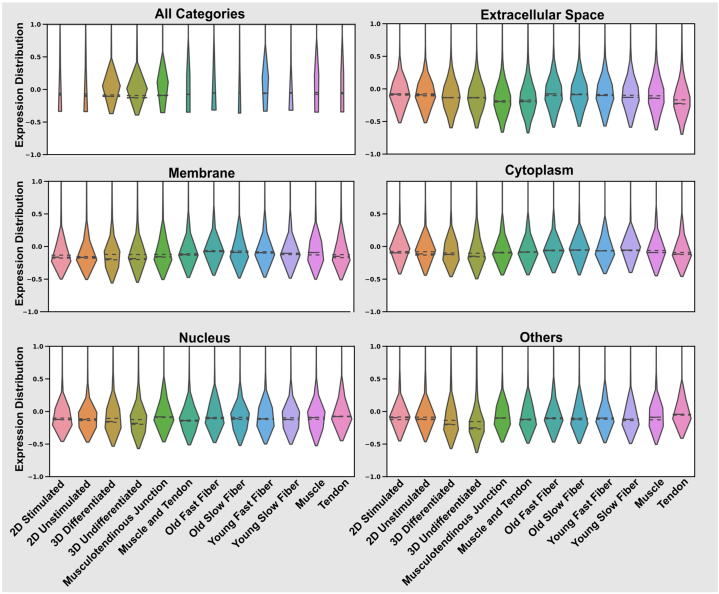
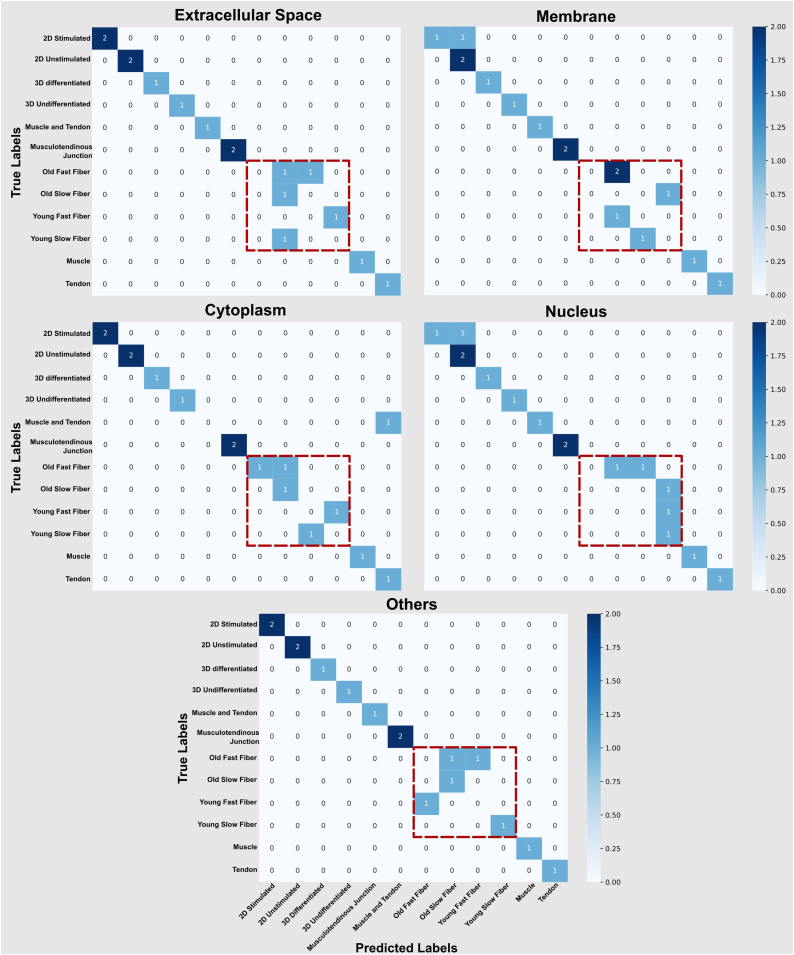
Omics techniques, such as proteomics, contain crucial data for understanding biological processes, but they remain underutilized due to their high dimensionality. Typically, proteomics research focuses narrowly on using a limited number of datasets, hindering cross-study comparisons, a problem that can potentially be addressed by machine learning. Despite this potential, machine learning has seen limited adoption in the field of proteomics. Here, skeletal muscle proteomics datasets from five separate studies were combined. These studies included conditions such as models (both 2D and 3D), skeletal muscle tissue, and adjacent tissues such as tendons. The collected data was preprocessed using MaxQuant, and then enriched using a Python script fetching structural and compositional details from UniProt and Ensembl databases. This was used to handle high-dimensional and sparsely labeled dataset by breaking it down into five smaller categories using cellular composition information and then training a Random Forest model for each category separately. Using biological context for interpreting the data resulted in improved model performance and made tailored analysis possible by reducing the dimensionality and increasing signal-to-noise ratio as well as only preserving biologically relevant features in each category. This integration of domain knowledge into data analysis and model training facilitated the discovery of new patterns while ensuring the retention of critical details, often overlooked when blind feature selection methods are used to exclude proteins with minimal expressions or variances. This approach was shown to be suitable for performing diverse analyses on individual as well as combined datasets within a broader biological context, ultimately leading to the identification of biologically relevant patterns. Besides from generating new biological insights, this approach can be used to perform tasks such as biomarker discovery, cluster analysis, classification, and anomaly detection more accurately, but incorporation of more datasets is needed to further expand the computational capabilities of such models in clinical settings.
组学技术,如蛋白质组学,包含理解生物过程的关键数据,但由于其高维度性,这些数据仍未得到充分利用。通常,蛋白质组学研究狭隘地集中于使用有限数量的数据集,这阻碍了跨研究比较,而机器学习可能可以解决这个问题。尽管有这种潜力,但机器学习在蛋白质组学领域的应用仍然有限。在这里,来自五项独立研究的骨骼肌蛋白质组学数据集被合并。这些研究包括诸如模型(二维和三维)、骨骼肌组织以及诸如肌腱等相邻组织等条件。收集到的数据使用MaxQuant进行预处理,然后使用一个Python脚本从UniProt和Ensembl数据库获取结构和组成细节进行富集。这被用于通过利用细胞组成信息将高维度且标记稀疏的数据集分解为五个较小的类别,然后分别为每个类别训练一个随机森林模型来处理该数据集。利用生物学背景来解释数据提高了模型性能,并通过降低维度、增加信噪比以及仅保留每个类别中生物学相关特征,使得定制分析成为可能。将领域知识整合到数据分析和模型训练中有助于发现新的模式,同时确保保留关键细节,而当使用盲目特征选择方法排除低表达或低方差蛋白质时,这些细节常常被忽视。这种方法被证明适用于在更广泛的生物学背景下对单个以及组合数据集进行各种分析,最终导致识别出生物学相关模式。除了产生新的生物学见解外,这种方法还可用于更准确地执行诸如生物标志物发现、聚类分析、分类和异常检测等任务,但需要纳入更多数据集以进一步扩展此类模型在临床环境中的计算能力。