Buncha Vadym, Lang Liwei, Fopiano Katie Anne, Ilatovskaya Daria V, Kapuku Gaston, Verin Alexander D, Bagi Zsolt
Department of Physiology, Medical College of Georgia, Augusta University, Augusta, Georgia, United States.
Department of Medicine, Georgia Prevention Institute, Medical College of Georgia, Augusta University, Augusta, Georgia, United States.
Am J Physiol Heart Circ Physiol. 2025 Feb 1;328(2):H283-H293. doi: 10.1152/ajpheart.00593.2024. Epub 2024 Dec 31.
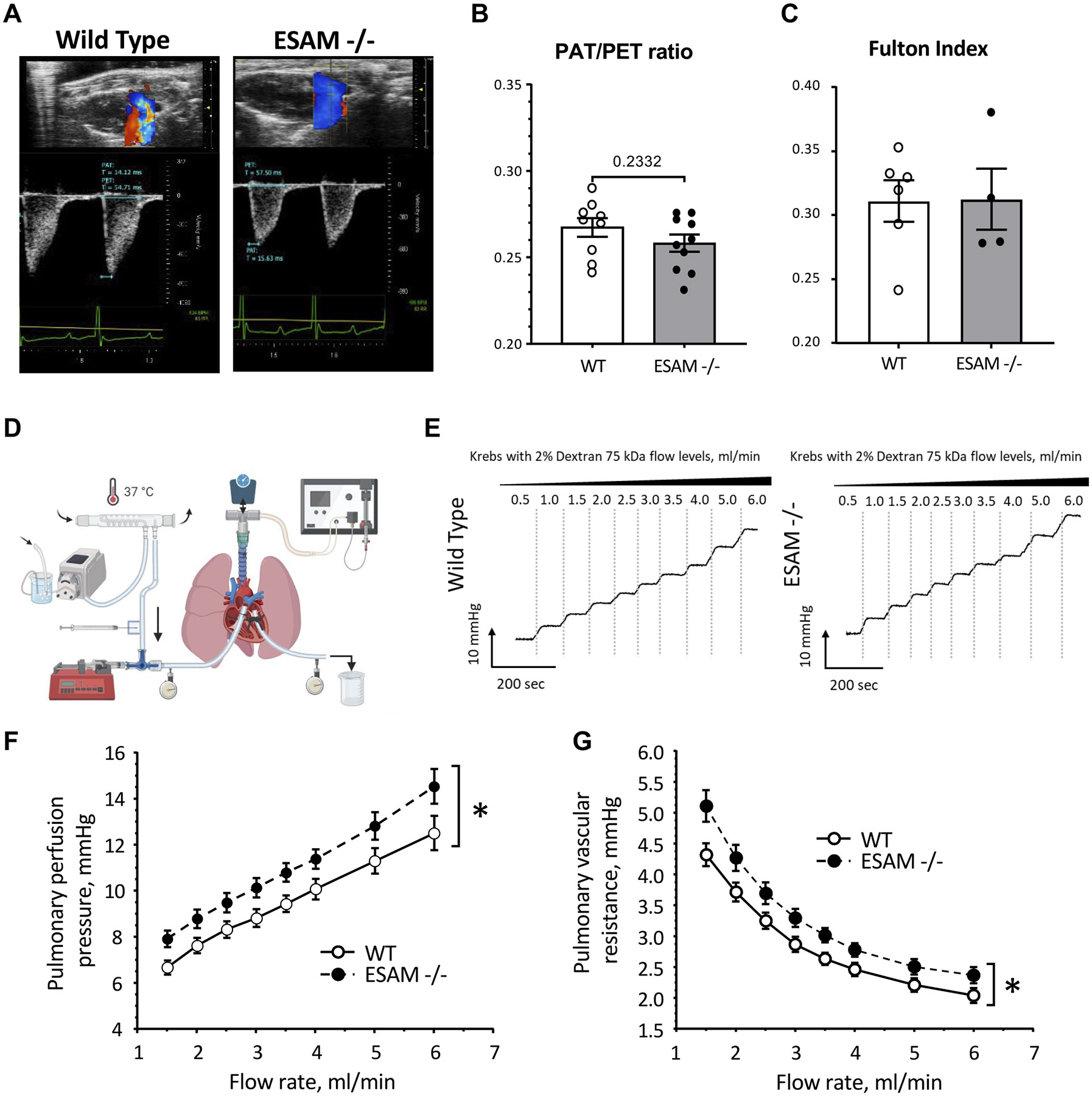
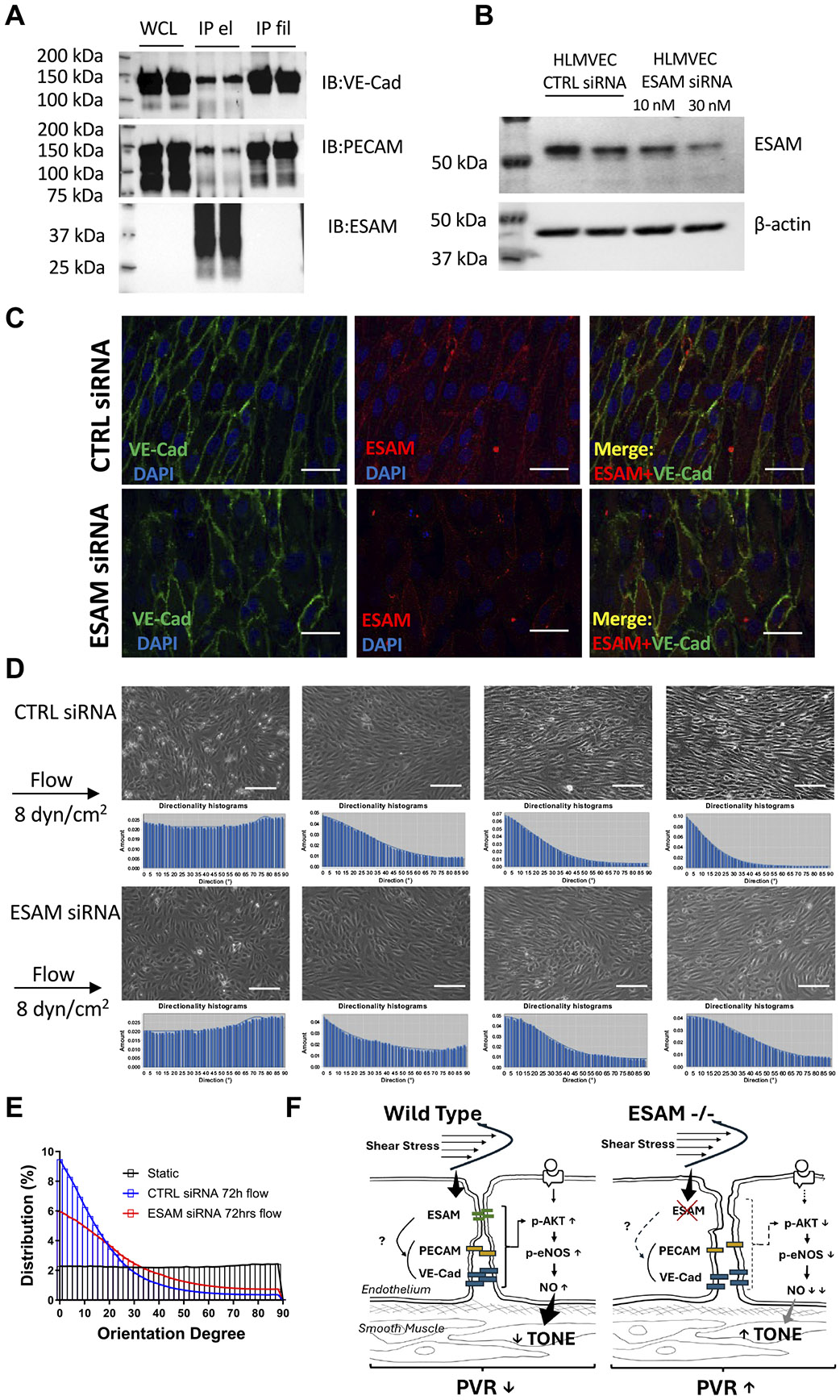
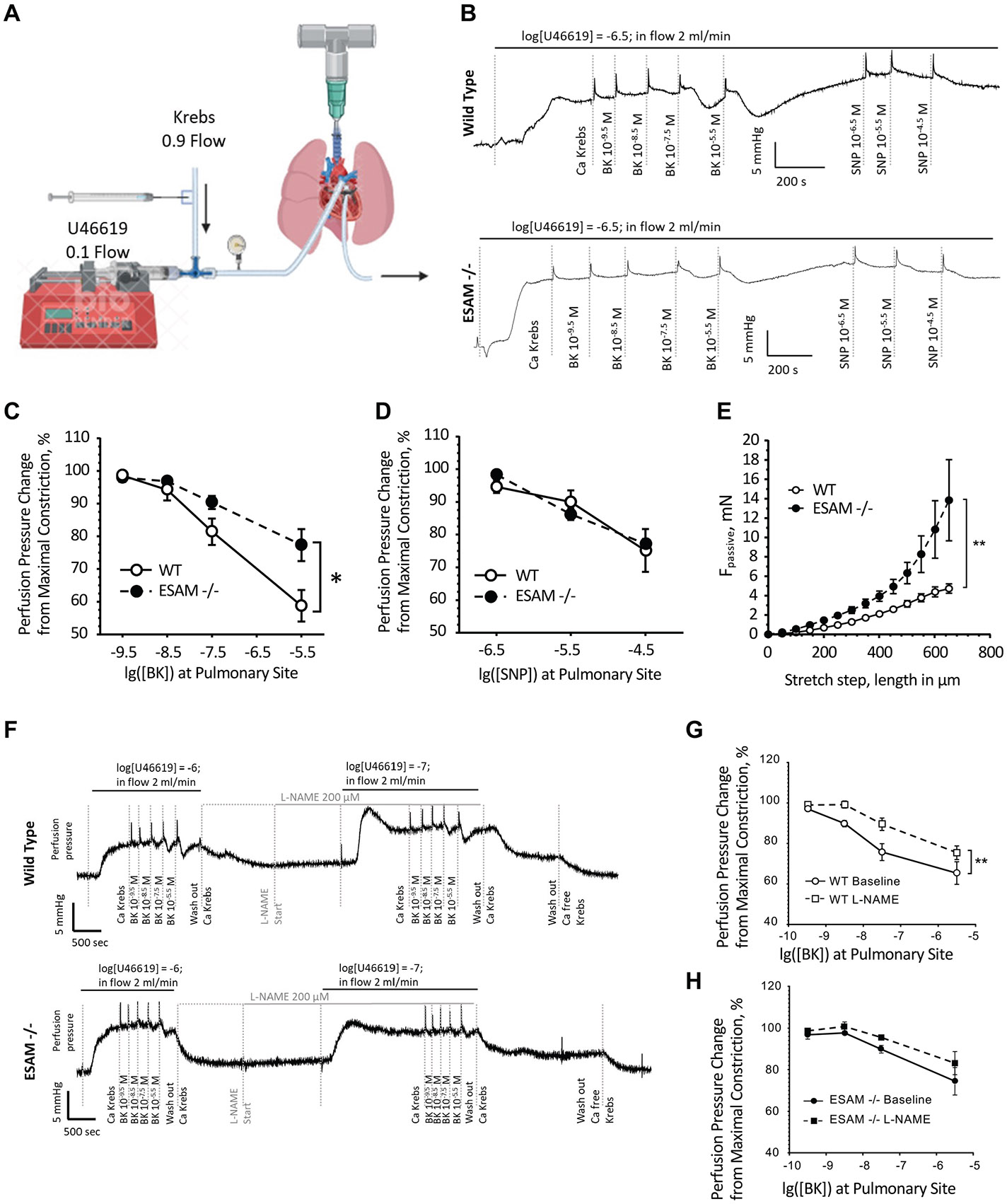
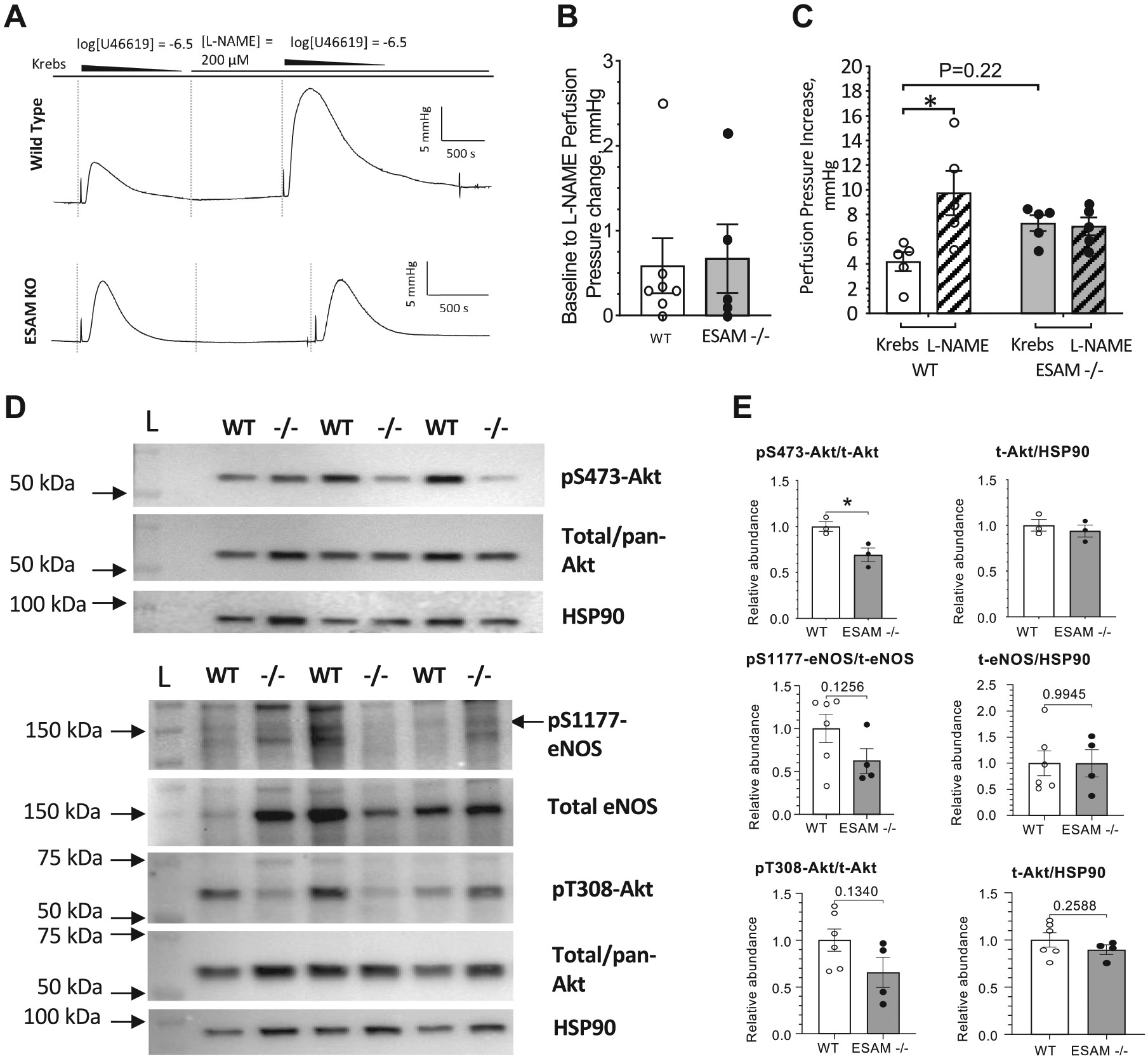
Endothelial cell-selective adhesion molecule (ESAM) is a member of tight junction molecules, highly abundant in the heart and the lung, and plays a role in regulating endothelial cell permeability. We previously reported that mice with genetic ESAM deficiency () exhibit coronary microvascular dysfunction leading to the development of left ventricular diastolic dysfunction. Here, we hypothesize that mice display impairments in the pulmonary vasculature, affecting the overall pulmonary vascular resistance (PVR). We utilized mice and employed isolated, ventilated, and perfused whole lung preparation to assess PVR independently of cardiac function. PVR was assessed in response to stepwise increases in flow, and also in response to perfusion of the endothelium-dependent agonist, bradykinin, the thromboxane analog, U46619, and the nitric oxide (NO) donor sodium nitroprusside (SNP). We found that PVR, at every applied flow rate, is significantly elevated in mice compared with WT mice. Bradykinin-induced reduction in PVR and U46619-induced increase in PVR were both diminished in mice, whereas SNP-induced responses were similar in wild-type (WT) and mice. Inhibition of NO synthase with (ω)-nitro-l-arginine methyl ester increased agonist-induced PVR in WT but not in mice. Pulmonary arteries isolated from mice exhibited a reduced level of phospho-Ser473-Akt and phospho-Ser1177-eNOS. Furthermore, in human lung microvascular endothelial cells cultured under flow conditions, we found that siRNA-mediated knockdown of ESAM impaired fluid shear stress-induced endothelial cell alignment. Thus, we suggest that ESAM plays an important role in the endothelium-dependent, flow/shear stress- and vasoactive agonist-stimulated, and NO-mediated maintenance of PVR in mice. Our study reveals a novel role for ESAM in contributing to the maintenance of pulmonary vascular resistance under normal physiological conditions. Employing mice with global genetic deficiency of ESAM and using isolated whole lung preparation, we show significant impairments in nitric oxide-mediated pulmonary artery function. In vitro cell culture studies demonstrate impaired fluid shear stress-induced cell alignment in human lung endothelial cells after siRNA-mediated ESAM knockdown.
内皮细胞选择性黏附分子(ESAM)是紧密连接分子的一员,在心脏和肺中高度丰富,并在调节内皮细胞通透性方面发挥作用。我们之前报道,基因敲除ESAM的小鼠()表现出冠状动脉微血管功能障碍,导致左心室舒张功能障碍的发生。在此,我们假设基因敲除ESAM的小鼠肺血管存在功能障碍,影响整体肺血管阻力(PVR)。我们使用基因敲除ESAM的小鼠,并采用离体、通气和灌注的全肺制备方法,独立于心脏功能来评估PVR。通过逐步增加流量以及灌注内皮依赖性激动剂缓激肽、血栓素类似物U46619和一氧化氮(NO)供体硝普钠(SNP)来评估PVR。我们发现,与野生型小鼠相比,在每个应用的流速下,基因敲除ESAM的小鼠的PVR均显著升高。缓激肽诱导的PVR降低和U46619诱导的PVR升高在基因敲除ESAM的小鼠中均减弱,而SNP诱导的反应在野生型(WT)小鼠和基因敲除ESAM的小鼠中相似。用(ω)-硝基-L-精氨酸甲酯抑制一氧化氮合酶可增加野生型小鼠激动剂诱导的PVR,但在基因敲除ESAM的小鼠中则不然。从基因敲除ESAM的小鼠分离的肺动脉显示磷酸化丝氨酸473-Akt和磷酸化丝氨酸1177-eNOS水平降低。此外,在流动条件下培养的人肺微血管内皮细胞中,我们发现siRNA介导的ESAM敲低损害了流体剪切应力诱导的内皮细胞排列。因此,我们认为ESAM在小鼠内皮依赖性、流动/剪切应力和血管活性激动剂刺激以及NO介导的PVR维持中起重要作用。我们的研究揭示了ESAM在正常生理条件下对维持肺血管阻力的新作用。通过使用ESAM全球基因敲除小鼠并采用离体全肺制备方法,我们显示一氧化氮介导的肺动脉功能存在显著障碍。体外细胞培养研究表明,siRNA介导的ESAM敲低后人肺内皮细胞中流体剪切应力诱导的细胞排列受损。