Li Jia-Peng, Qiu Shu, Tai Guang-Jie, Liu Yi-Ming, Wei Wei, Fu Meng-Meng, Fang Pan-Qi, Otieno Joseph Nicolao, Battulga Tungalag, Li Xiao-Xue, Xu Ming
Department of Clinical Pharmacy, School of Preclinical Medicine and Clinical Pharmacy, China Pharmaceutical University, 24 Tong jia Lane, Nanjing, 210009, People's Republic of China.
Department of Thoracic and Cardiovascular Surgery, Nanjing First Hospital, Nanjing Medical University, Nanjing, 210009, People's Republic of China.
Cardiovasc Diabetol. 2025 Jan 6;24(1):6. doi: 10.1186/s12933-024-02541-3.
Inflammatory diseases impair the reparative properties of endothelial progenitor cells (EPC); however, the involvement of diabetes in EPC dysfunction associated with myocardial infarction (MI) remains unknown.
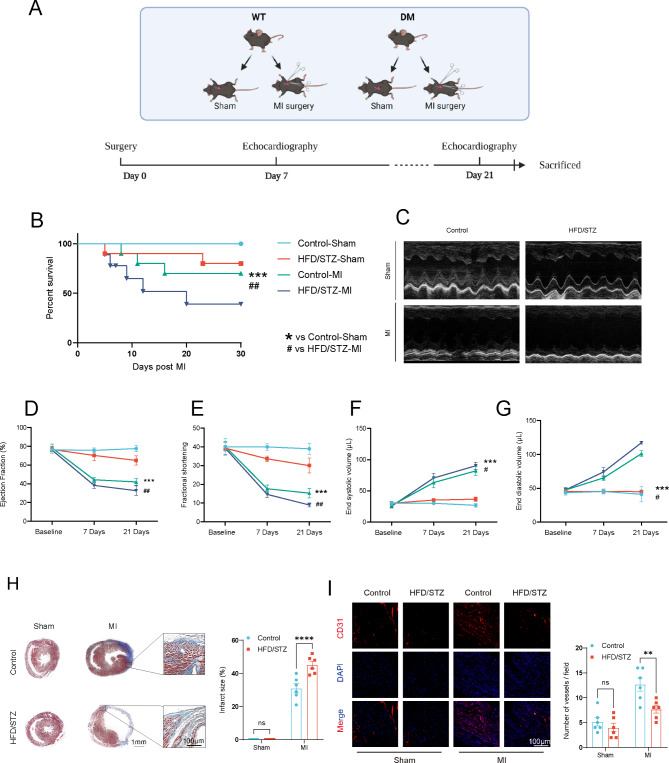
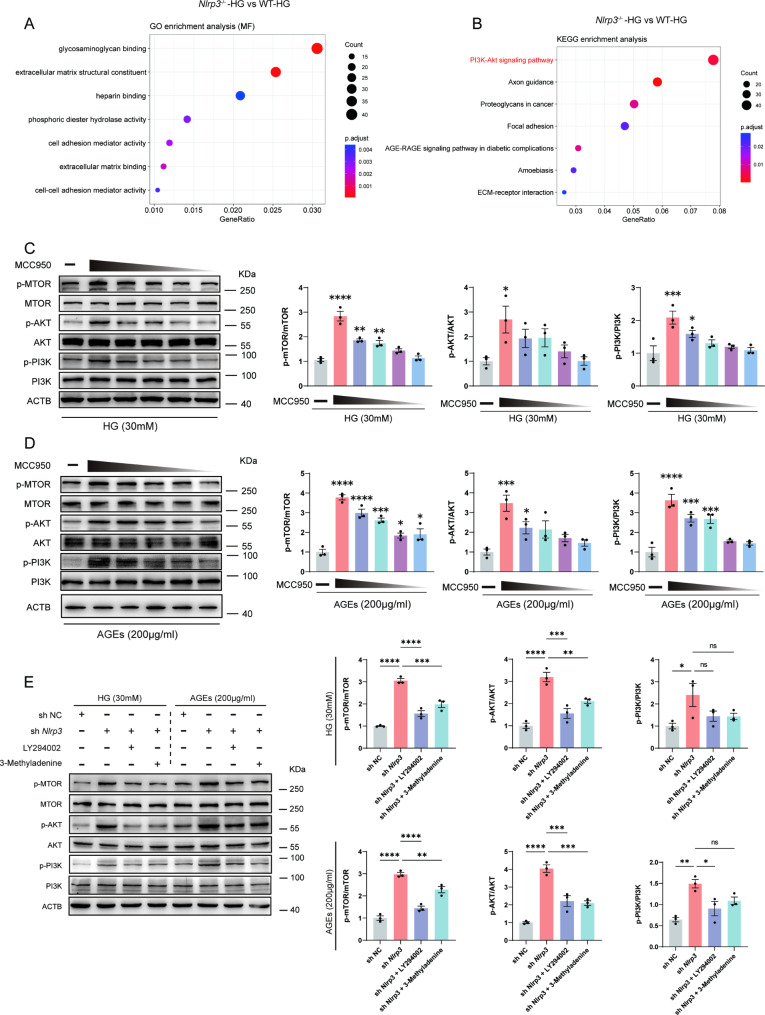
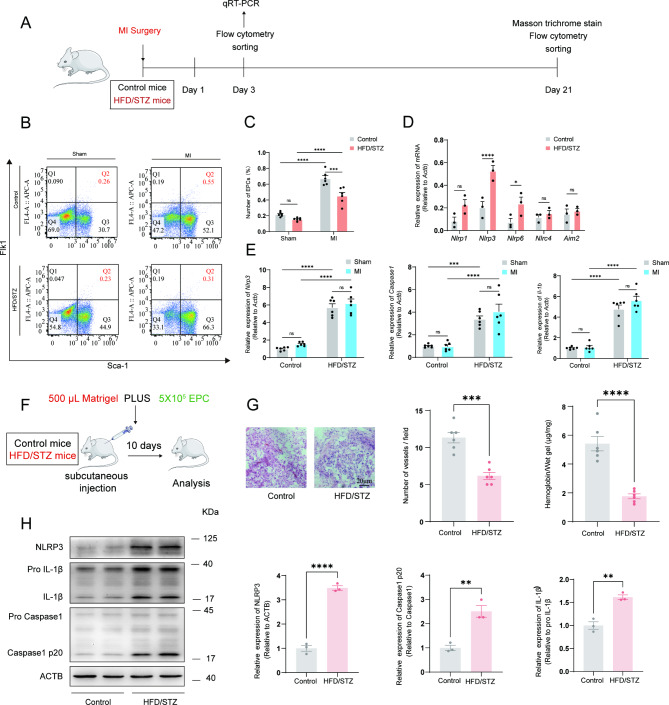
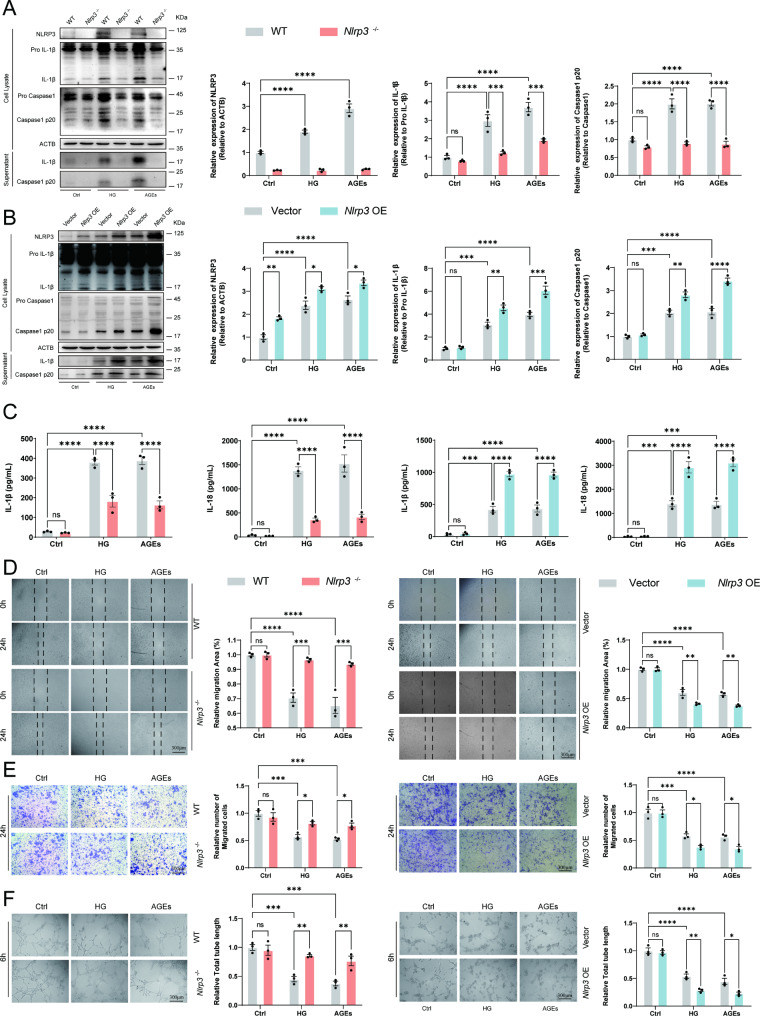
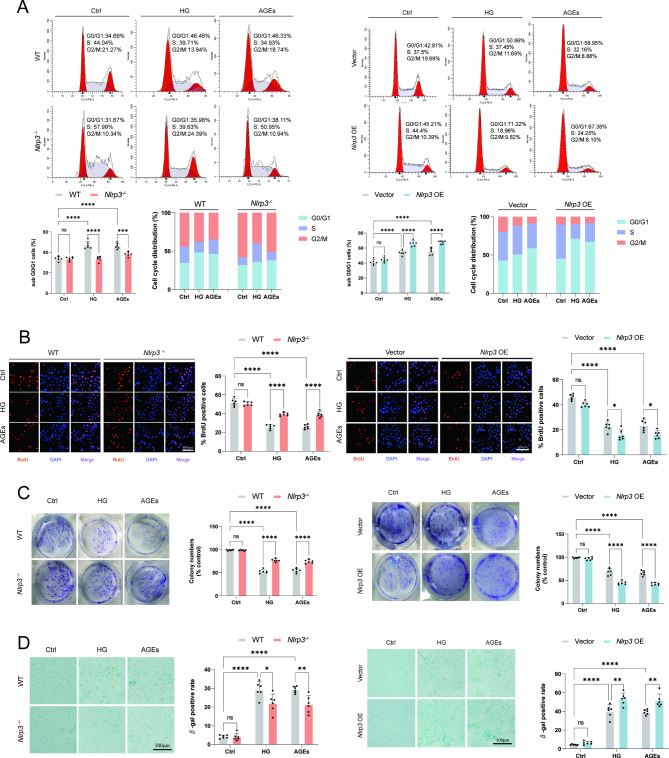
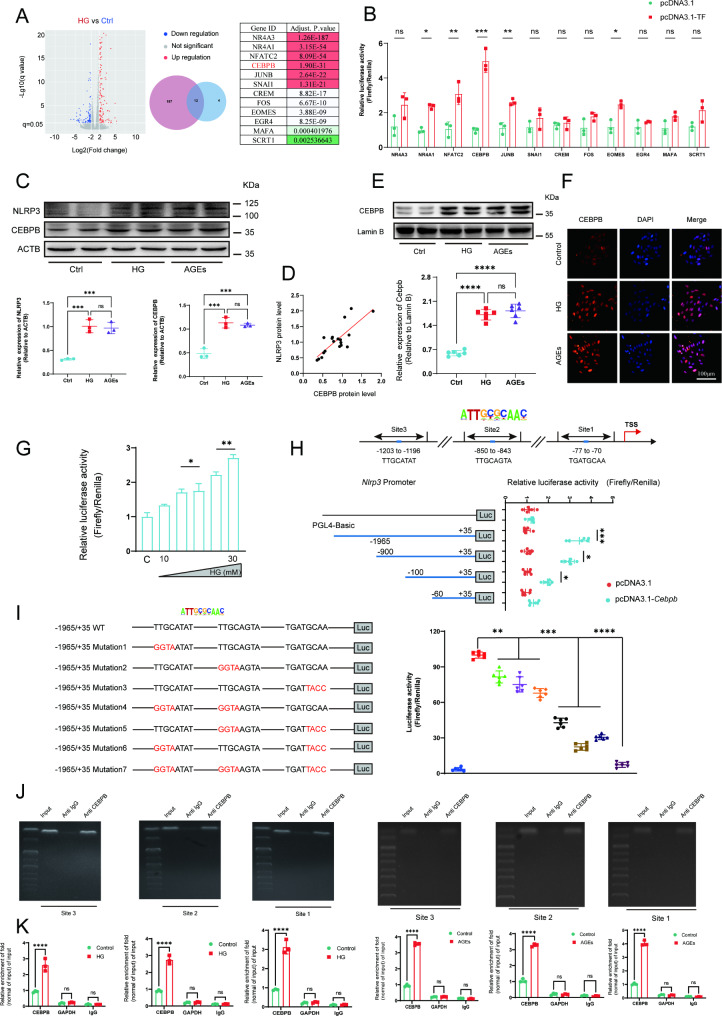
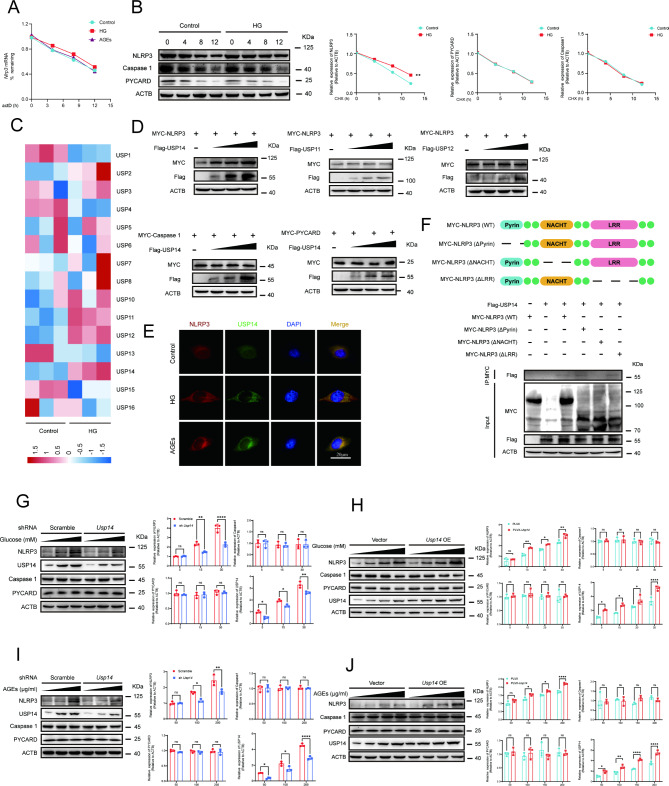
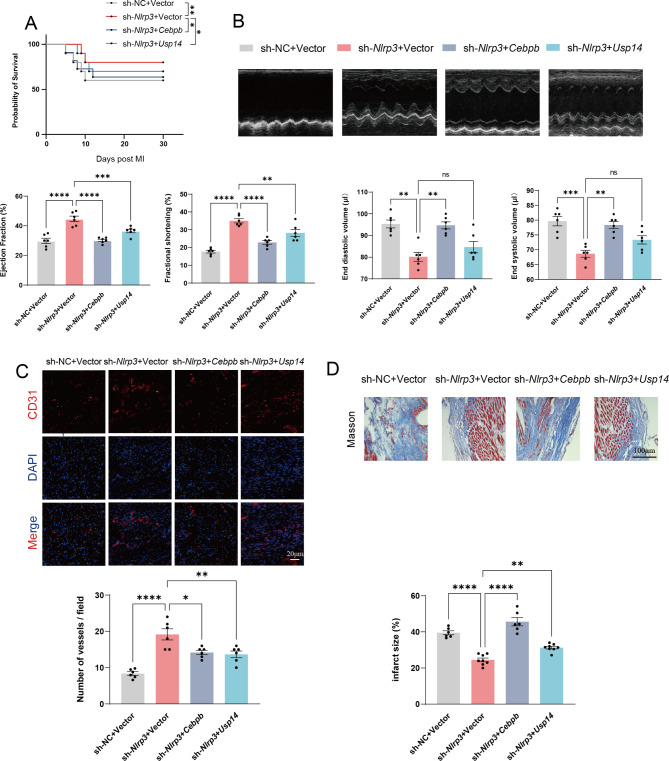
A model was established combining high-fat diet (HFD)/streptozotocin (STZ)-induced diabetic mice with myocardial infarction. The therapeutic effects of transplanted wild-type EPC, Nlrp3 knockout EPC, and Nlrp3 overexpression EPC were evaluated. Chip and Luciferase assay revealed CEBPB regulated the transcriptional expression of Nlrp3 as a transcription factor in EPC stimulated by high glucose (HG) or advanced glycation end products (AGEs). CO-IP results suggested that USP14 selectively suppressed NLRP3 degradation. KEGG enrichment revealed PI3K/ Akt/mTOR signaling showed striking significance in the entire pathway.
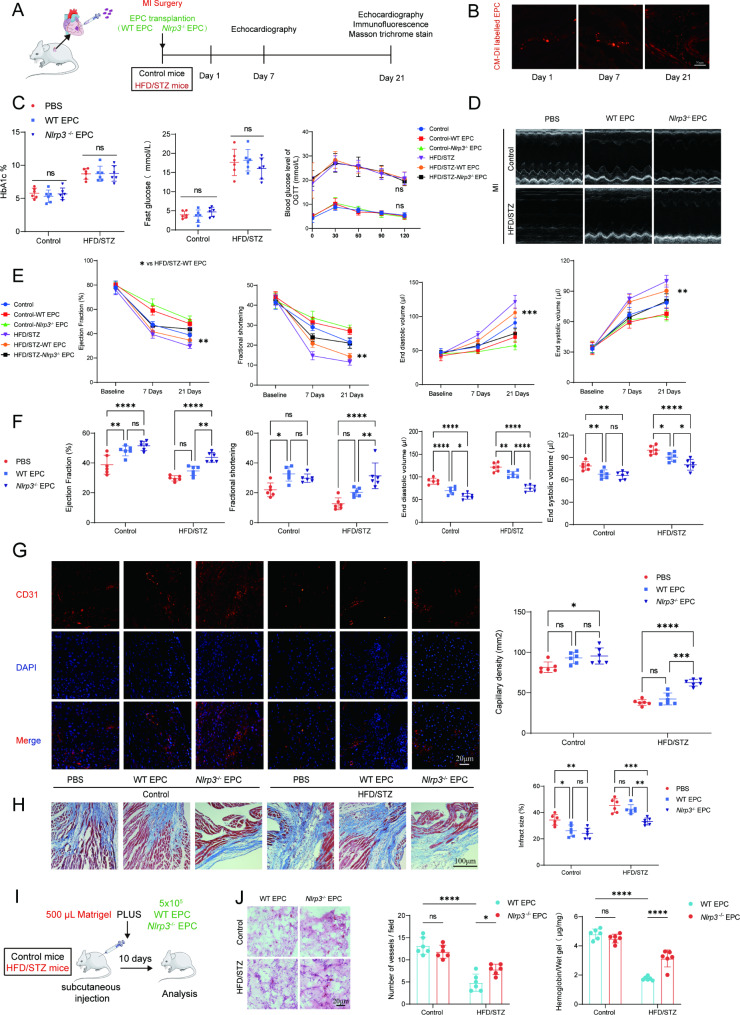
In our study, wild-type, Nlrp3 knockout and Nlrp3 overexpressed EPC, intracardiac injections effectively improved cardiac function, increased angiogenesis, and reduced infarct size in mice with myocardial infarction. However, in the HFD/STZ-induced diabetic mice model combined with myocardial infarction, Nlrp3 knockout EPC significantly restored angiogenic capacity. Mechanically, CEBPB regulated the transcriptional level of Nlrp3 as a transcription factor in EPC. Meanwhile, we found that USP14 selectively suppressed NLRP3 protein degradation through the USP motif on the NACHT domain in mediating inflammasome activation. Cardiac functional outcomes in recipient mice after intramyocardial injection of shNlrp3 EPC overexpressing CEBPB or USP14 validated the modulation of EPC function by regulating Nlrp3 transcription or post-translational modification. Furthermore, KEGG enrichment and validation at the protein levels revealed PI3K/ Akt/mTOR cascade might be a downstream signal for NLRP3 inflammasome.
Our study provides a new understanding of how diabetes affected progenitor cell-mediated cardiac repair and identifies NLRP3 as a new therapeutic target for improving myocardial infarction repair in inflammatory diseases.
炎症性疾病会损害内皮祖细胞(EPC)的修复特性;然而,糖尿病在与心肌梗死(MI)相关的EPC功能障碍中的作用仍不清楚。
建立了高脂饮食(HFD)/链脲佐菌素(STZ)诱导的糖尿病小鼠与心肌梗死相结合的模型。评估了移植野生型EPC、Nlrp3基因敲除EPC和Nlrp3过表达EPC的治疗效果。芯片和荧光素酶测定显示,在高糖(HG)或晚期糖基化终产物(AGEs)刺激的EPC中,CEBPB作为转录因子调节Nlrp3的转录表达。免疫共沉淀结果表明,USP14选择性抑制NLRP3的降解。KEGG富集分析显示PI3K/Akt/mTOR信号通路在整个信号通路中具有显著意义。
在我们的研究中,野生型、Nlrp3基因敲除和Nlrp3过表达的EPC经心内注射可有效改善心肌梗死小鼠的心脏功能,增加血管生成,并减小梗死面积。然而,在HFD/STZ诱导的糖尿病小鼠合并心肌梗死模型中,Nlrp3基因敲除的EPC显著恢复了血管生成能力。从机制上讲,CEBPB作为EPC中的转录因子调节Nlrp3的转录水平。同时,我们发现USP14通过NACHT结构域上的USP基序选择性抑制NLRP3蛋白降解,从而介导炎性小体激活。心肌内注射过表达CEBPB或USP14的shNlrp3 EPC后,受体小鼠的心脏功能结果证实了通过调节Nlrp3转录或翻译后修饰对EPC功能的调节作用。此外,KEGG富集分析和蛋白质水平验证显示PI3K/Akt/mTOR级联可能是NLRP3炎性小体的下游信号。
我们的研究为糖尿病如何影响祖细胞介导的心脏修复提供了新的认识,并确定NLRP3是改善炎症性疾病心肌梗死修复的新治疗靶点。