Sharma Ganesh, Kumar Neeraj, Sharma Chandra Shekhar, Alqahtani Taha, Tiruneh Yewulsew Kebede, Sultana Sharifa, Rolim Silva Gabriel Vinícius, de Lima Menezes Gabriela, Zaki Magdi E A, Nobre Oliveira Jonas Ivan
Department of Pharmaceutical Chemistry, Bhupal Nobles' College of Pharmacy, Bhupal Nobles' University, Udaipur, 313002, India.
Department of Pharmacology, College of Pharmacy, King Khalid University, Abha, 62529, Saudi Arabia.
Sci Rep. 2025 Jan 22;15(1):2830. doi: 10.1038/s41598-025-86016-9.
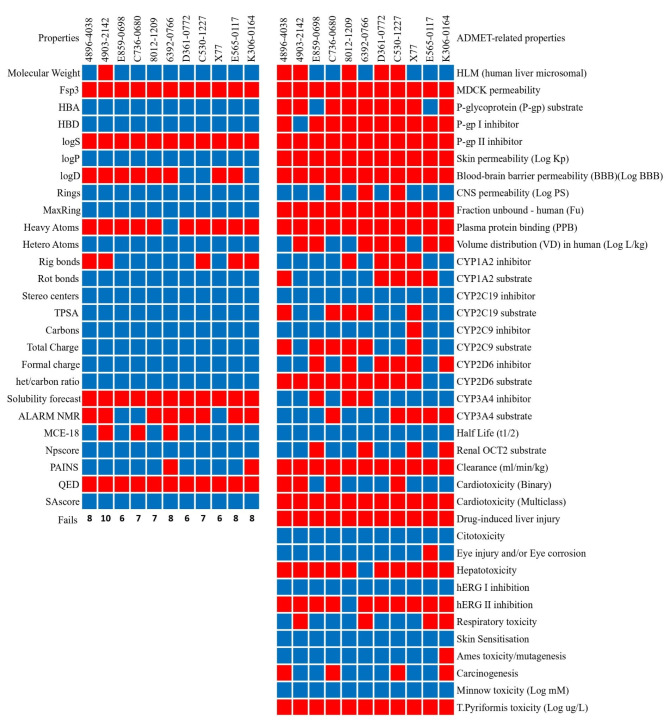
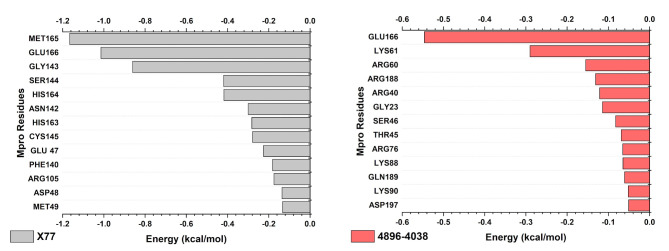
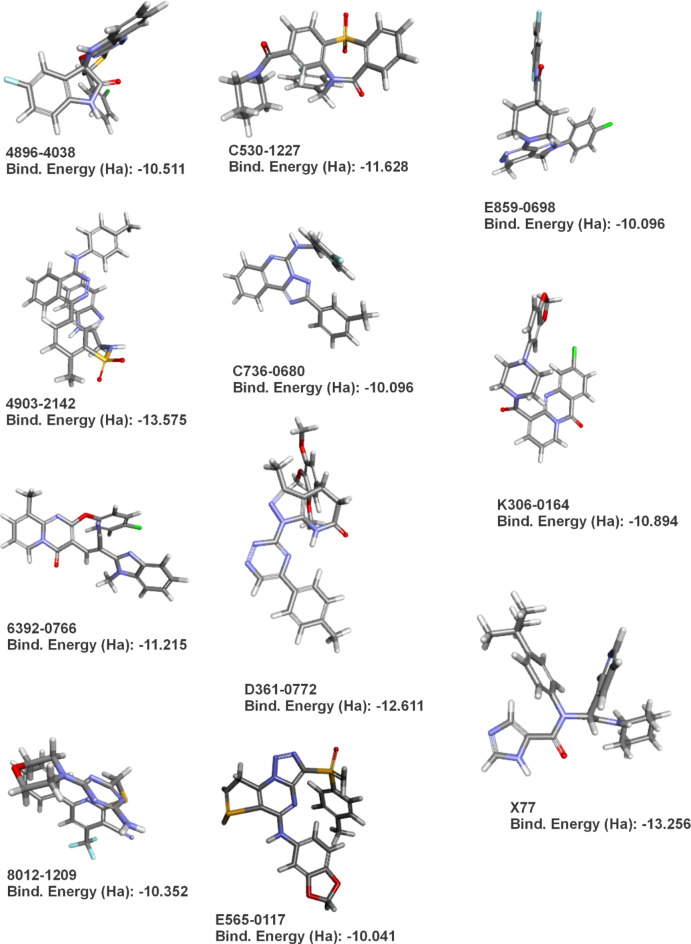
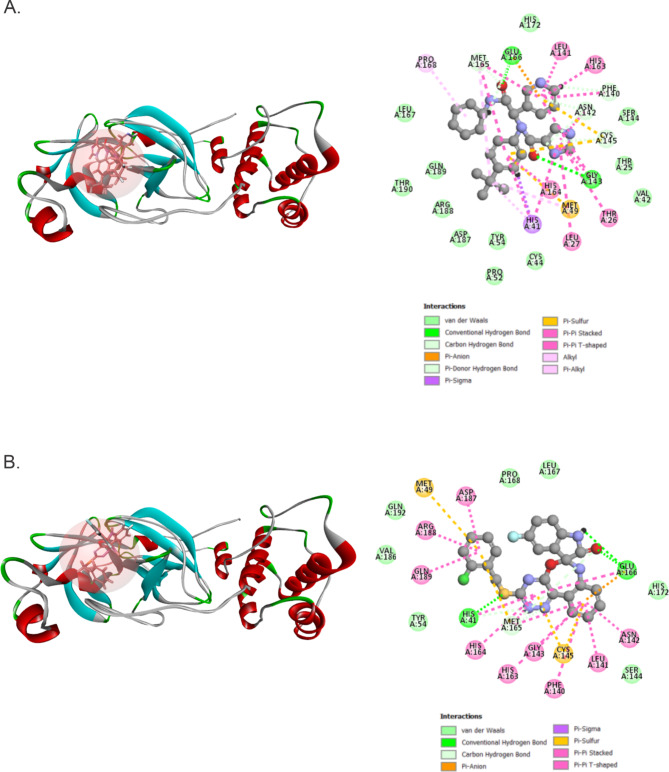
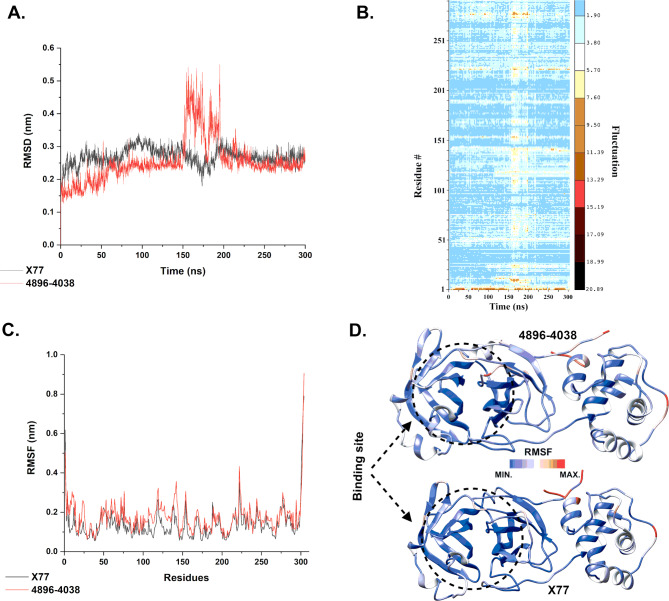
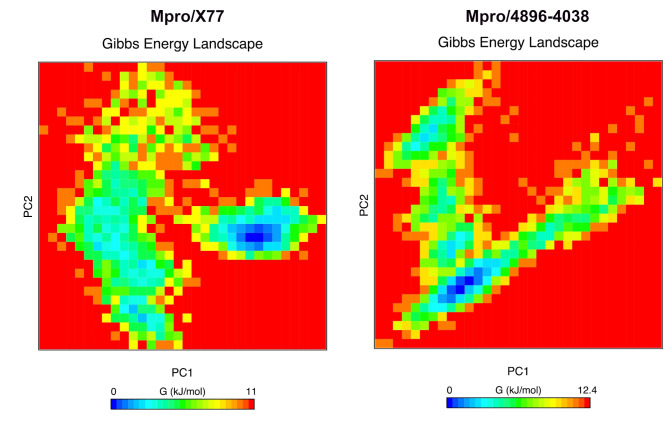
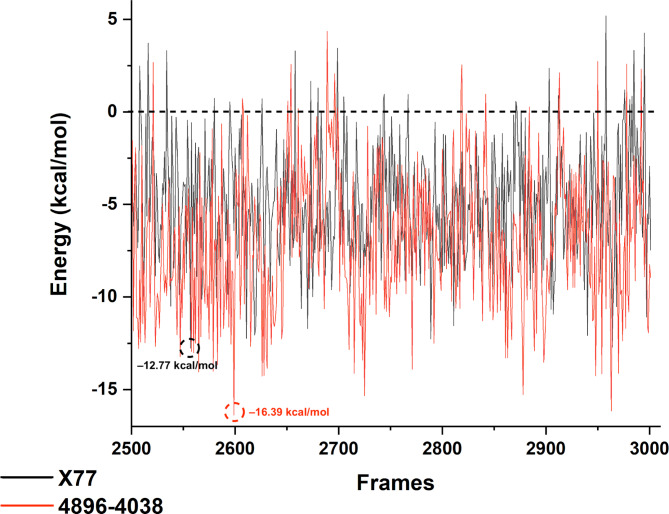
The COVID-19 pandemic caused by SARS-CoV-2 continues to pose a major challenge to global health. Targeting the main protease of the virus (Mpro), which is essential for viral replication and transcription, offers a promising approach for therapeutic intervention. In this study, advanced computational techniques such as molecular docking and molecular dynamics simulations were used to screen a series of antiviral compounds for their potential inhibitory effect on the SARS-CoV-2 Mpro. A comprehensive analysis of compounds from the ChemDiv and PubChem databases was performed. The physicochemical properties, pharmacokinetics, and ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) profiles were evaluated to determine drug similarity and safety. Compound 4896 - 4038 proved to be the most promising candidate. It exhibited a favorable balance between molecular weight (491.06) and lipophilicity (logP 3.957), high intestinal absorption (92.119%), and broad tissue distribution (VDss of 0.529), indicating good oral bioavailability and therapeutic potential. Molecular docking studies showed that 4896 - 4038 has a strong binding affinity to the active site of Mpro and forms key interactions, such as hydrogen bonds, carbon-hydrogen bonds, pi-sulfur, and multiple van der Waals and pi-pi stacked bonds. The binding energy was comparable to that of the reference drug X77, indicating potential efficacy. Molecular dynamics simulations over 300 ns confirmed the stability of the Mpro/4896 - 4038 complex of protein-ligand. Free energy landscape mapping and MM/PBSA calculations further substantiated the favorable binding and stability of the complex. Importantly, 4896 - 4038 exhibited a comparatively favorable safety profile. In summary, compound 4896 - 4038 shows significant potential as a potent SARS-CoV-2 Mpro inhibitor, combining potent inhibitory activity with favorable pharmacokinetic and safety profiles. These results support the further development of 4896 - 4038 as a promising therapeutic agent in the fight against COVID-19 that warrants experimental validation and clinical investigation.
由严重急性呼吸综合征冠状病毒2(SARS-CoV-2)引起的2019冠状病毒病(COVID-19)大流行继续对全球健康构成重大挑战。针对病毒的主要蛋白酶(Mpro),其对病毒复制和转录至关重要,为治疗干预提供了一种有前景的方法。在本研究中,使用了诸如分子对接和分子动力学模拟等先进的计算技术来筛选一系列抗病毒化合物对SARS-CoV-2 Mpro的潜在抑制作用。对来自ChemDiv和PubChem数据库的化合物进行了全面分析。评估了其物理化学性质、药代动力学以及吸收、分布、代谢、排泄和毒性(ADMET)特征,以确定药物相似性和安全性。化合物4896 - 4038被证明是最有前景的候选物。它在分子量(491.06)和亲脂性(logP 3.957)之间表现出良好的平衡,具有较高的肠道吸收(92.119%)和广泛的组织分布(稳态分布容积为0.529),表明具有良好的口服生物利用度和治疗潜力。分子对接研究表明,4896 - 4038与Mpro的活性位点具有很强的结合亲和力,并形成关键相互作用,如氢键、碳氢键、π-硫键以及多个范德华力和π-π堆积键。结合能与参考药物X77相当,表明具有潜在疗效。超过300纳秒的分子动力学模拟证实了蛋白质-配体的Mpro/4896 - 4038复合物的稳定性。自由能景观映射和MM/PBSA计算进一步证实了该复合物的良好结合和稳定性。重要的是,4896 - 4038表现出相对良好的安全性。总之,化合物4896 - 4038作为一种有效的SARS-CoV-2 Mpro抑制剂显示出巨大潜力,将强大的抑制活性与良好的药代动力学和安全性特征相结合。这些结果支持将4896 - 4038进一步开发为一种有前景的治疗剂,用于对抗COVID-19,这值得进行实验验证和临床研究。