Bioinformatics Laboratory, Department of Statistics, Faculty of Science, University of Rajshahi, Rajshahi 6205, Bangladesh.
Department of Chemistry, Faculty of Science, University of Rajshahi, Rajshahi 6205, Bangladesh.
Molecules. 2024 May 27;29(11):2524. doi: 10.3390/molecules29112524.
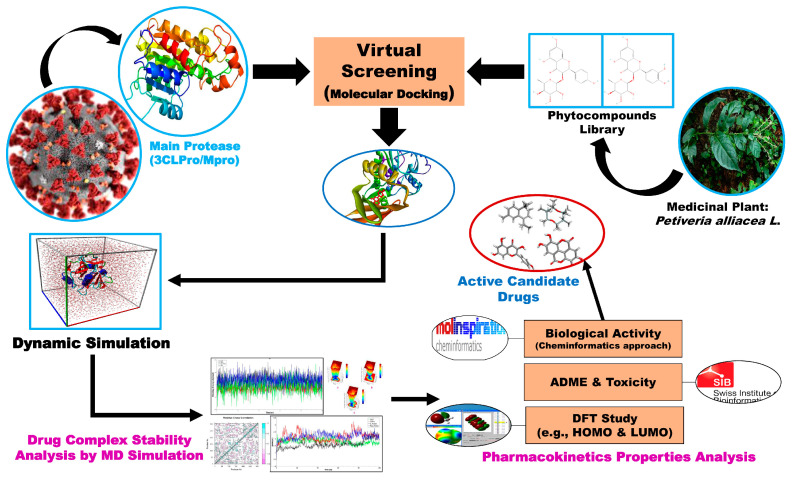
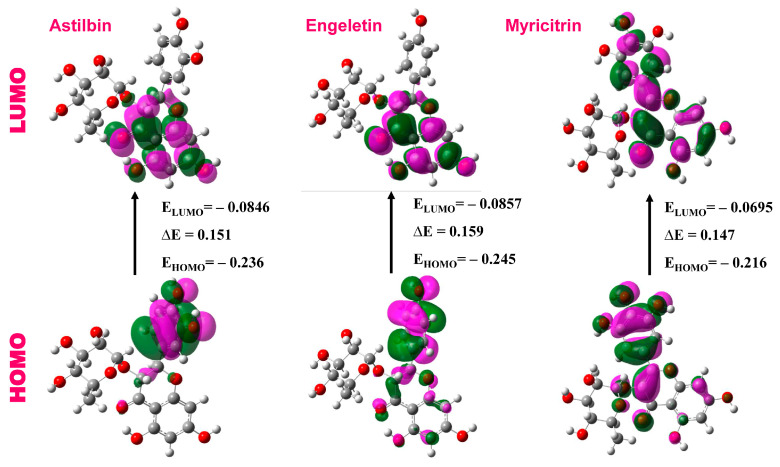
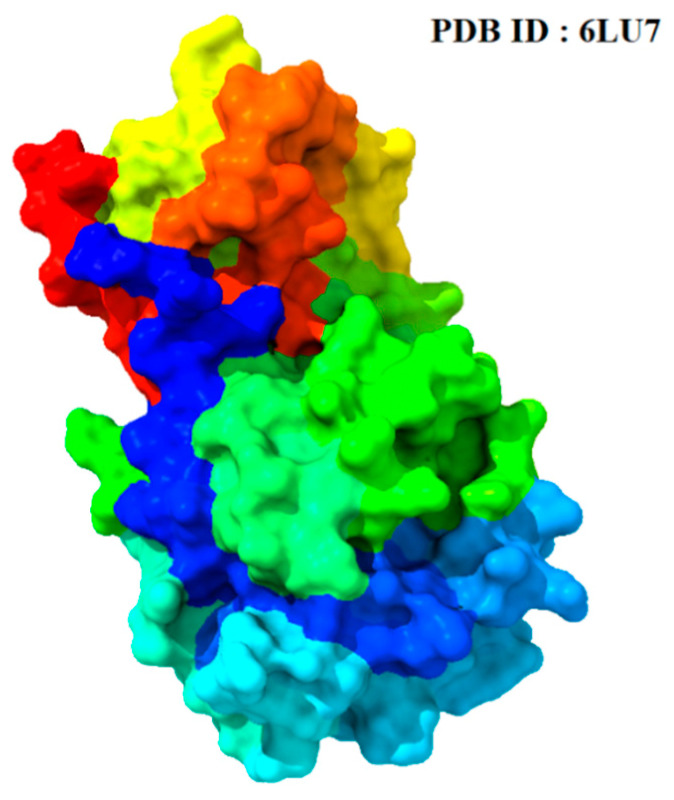
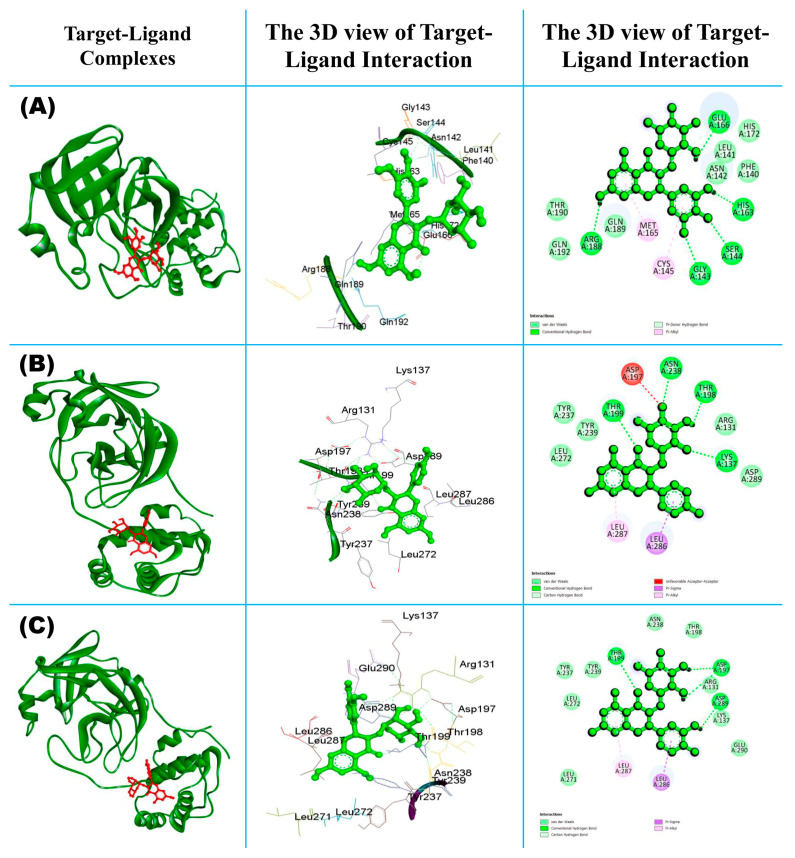
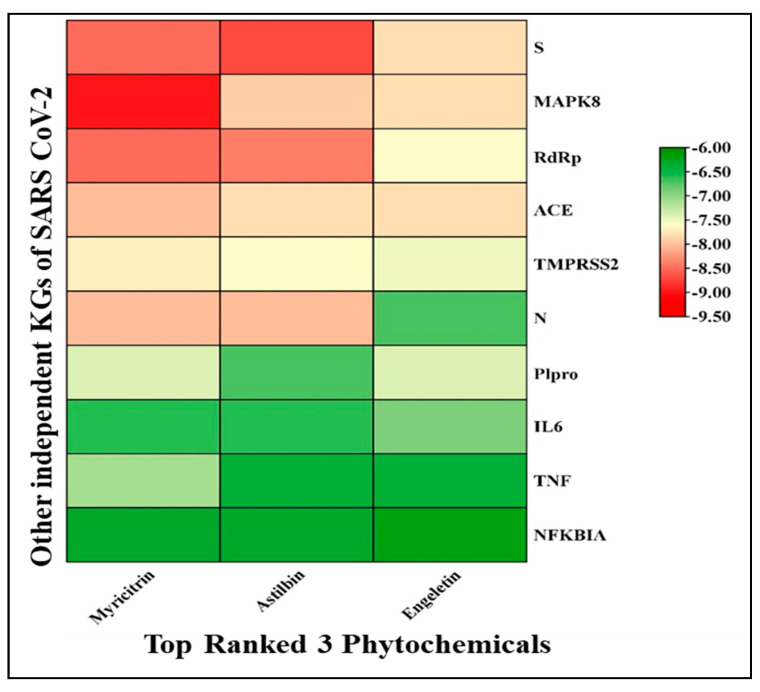
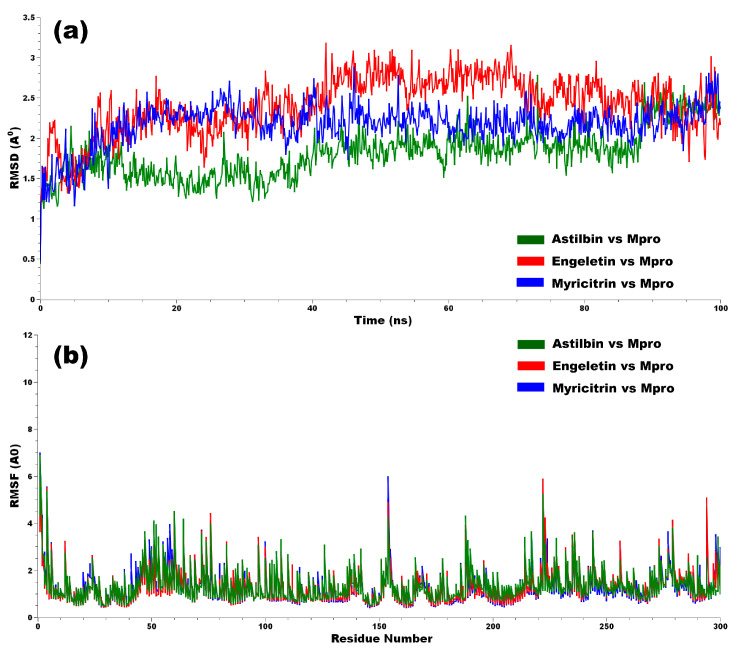
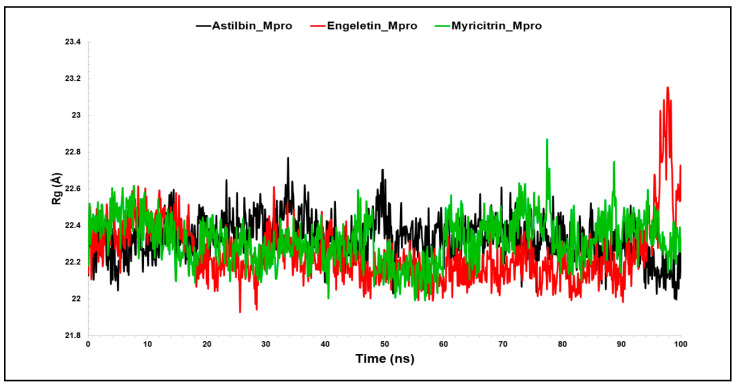
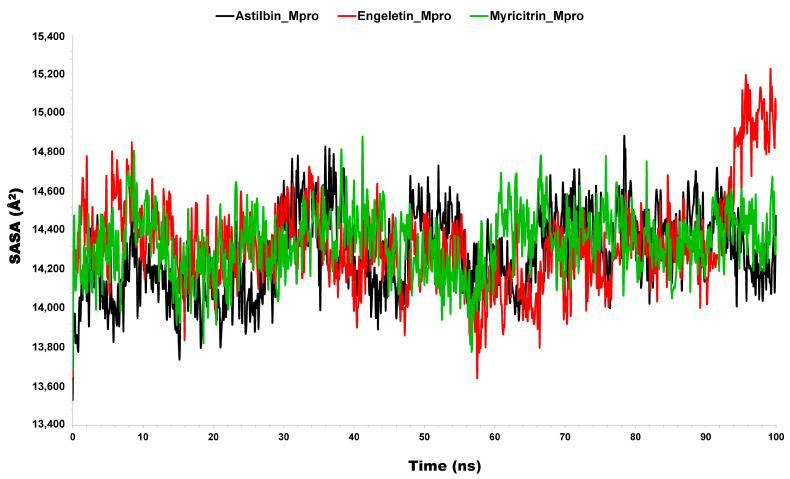
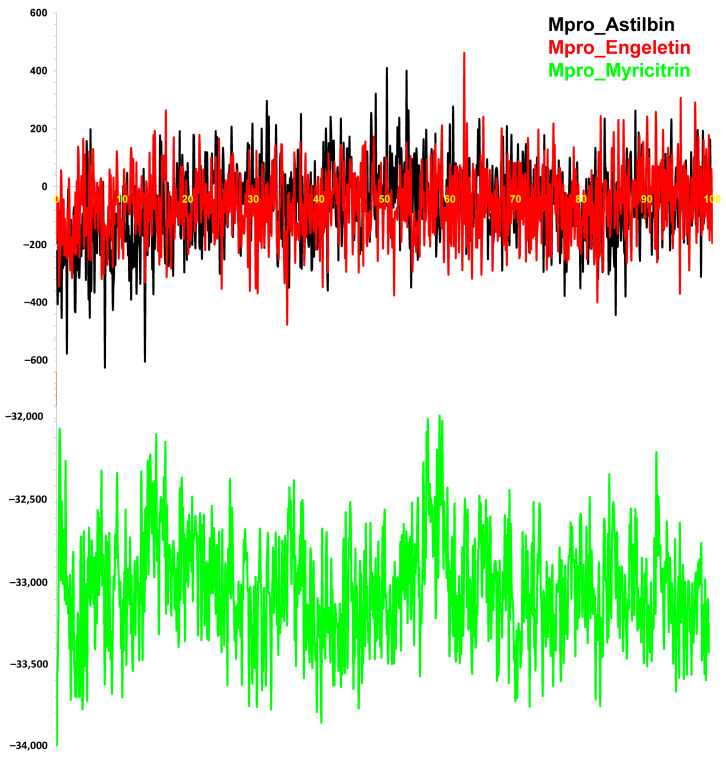
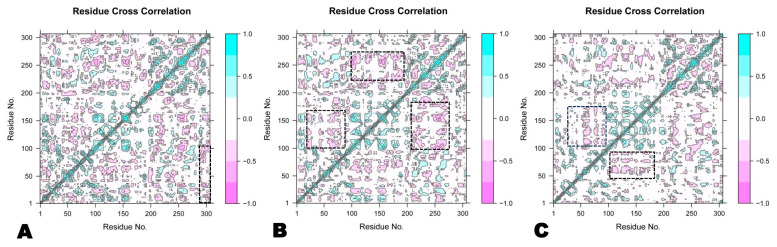
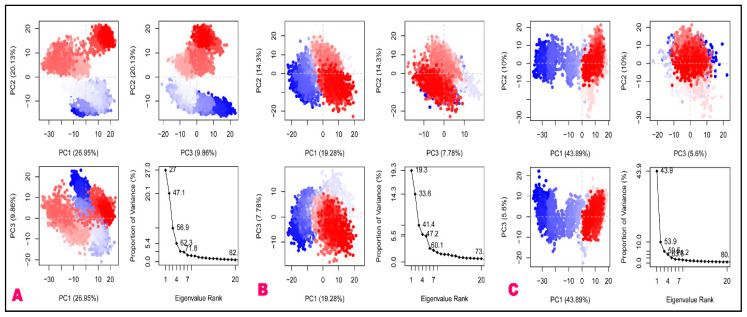
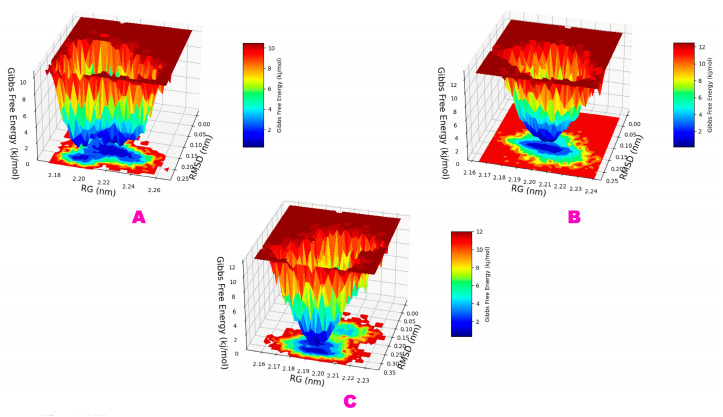
The outbreak of SARS-CoV-2, also known as the COVID-19 pandemic, is still a critical risk factor for both human life and the global economy. Although, several promising therapies have been introduced in the literature to inhibit SARS-CoV-2, most of them are synthetic drugs that may have some adverse effects on the human body. Therefore, the main objective of this study was to carry out an in-silico investigation into the medicinal properties of L. ( L.)-mediated phytocompounds for the treatment of SARS-CoV-2 infections since phytochemicals have fewer adverse effects compared to synthetic drugs. To explore potential phytocompounds from L. as candidate drug molecules, we selected the infection-causing main protease (Mpro) of SARS-CoV-2 as the receptor protein. The molecular docking analysis of these receptor proteins with the different phytocompounds of L. was performed using AutoDock Vina. Then, we selected the three top-ranked phytocompounds (myricitrin, engeletin, and astilbin) as the candidate drug molecules based on their highest binding affinity scores of -8.9, -8.7 and -8.3 (Kcal/mol), respectively. Then, a 100 ns molecular dynamics (MD) simulation study was performed for their complexes with Mpro using YASARA software, computed RMSD, RMSF, PCA, DCCM, MM/PBSA, and free energy landscape (FEL), and found their almost stable binding performance. In addition, biological activity, ADME/T, DFT, and drug-likeness analyses exhibited the suitable pharmacokinetics properties of the selected phytocompounds. Therefore, the results of this study might be a useful resource for formulating a safe treatment plan for SARS-CoV-2 infections after experimental validation in wet-lab and clinical trials.
新型冠状病毒(SARS-CoV-2)的爆发,也就是我们熟知的 COVID-19 大流行,仍然是人类生命和全球经济的重大风险因素。尽管文献中已经介绍了几种有前途的抑制 SARS-CoV-2 的疗法,但它们大多数是合成药物,可能对人体有一些不良反应。因此,本研究的主要目的是对 L.(李属)介导的植物化合物治疗 SARS-CoV-2 感染的药用特性进行计算机研究,因为与合成药物相比,植物化学物质的不良反应较少。为了从 L. 中探索潜在的植物化合物作为候选药物分子,我们选择 SARS-CoV-2 的感染主要蛋白酶(Mpro)作为受体蛋白。使用 AutoDock Vina 对这些受体蛋白与 L. 的不同植物化合物进行分子对接分析。然后,我们根据它们的最高结合亲和力评分(-8.9、-8.7 和-8.3(Kcal/mol)),选择了三种排名最高的植物化合物(杨梅素、茵陈素和槲皮素)作为候选药物分子。然后,使用 YASARA 软件对它们与 Mpro 的复合物进行了 100ns 的分子动力学(MD)模拟研究,计算了 RMSD、RMSF、PCA、DCCM、MM/PBSA 和自由能景观(FEL),发现它们具有几乎稳定的结合性能。此外,生物活性、ADME/T、DFT 和药物相似性分析表明所选植物化合物具有合适的药代动力学特性。因此,在进行湿实验室和临床试验的实验验证后,本研究的结果可能为制定 SARS-CoV-2 感染的安全治疗方案提供有用的资源。