Ogunyemi Oludare M, Gyebi Gideon A, Olawale Femi, Ibrahim Ibrahim M, Iwaloye Opeyemi, Fabusiwa Modupe M, Omowaye Stephen, Oloyede Omotade I, Olaiya Charles O
Structural and Computational Biology Group, Nutritional and Industrial Biochemistry Research Unit, Department of Biochemistry, College of Medicine, University of Ibadan, Ibadan 200005, Nigeria.
Department of Biochemistry, Faculty of Science and Technology, Bingham University, New Karu, Nasarawa 961105, Nigeria.
Bioinform Adv. 2024 Dec 19;5(1):vbae205. doi: 10.1093/bioadv/vbae205. eCollection 2025.
Investigating novel drug-target interactions is crucial for expanding the chemical space of emerging therapeutic targets in human diseases. Herein, we explored the interactions of dipeptidyl peptidase-4 and protein tyrosine phosphatase 1B with selected terpenoids from African antidiabetic plants.
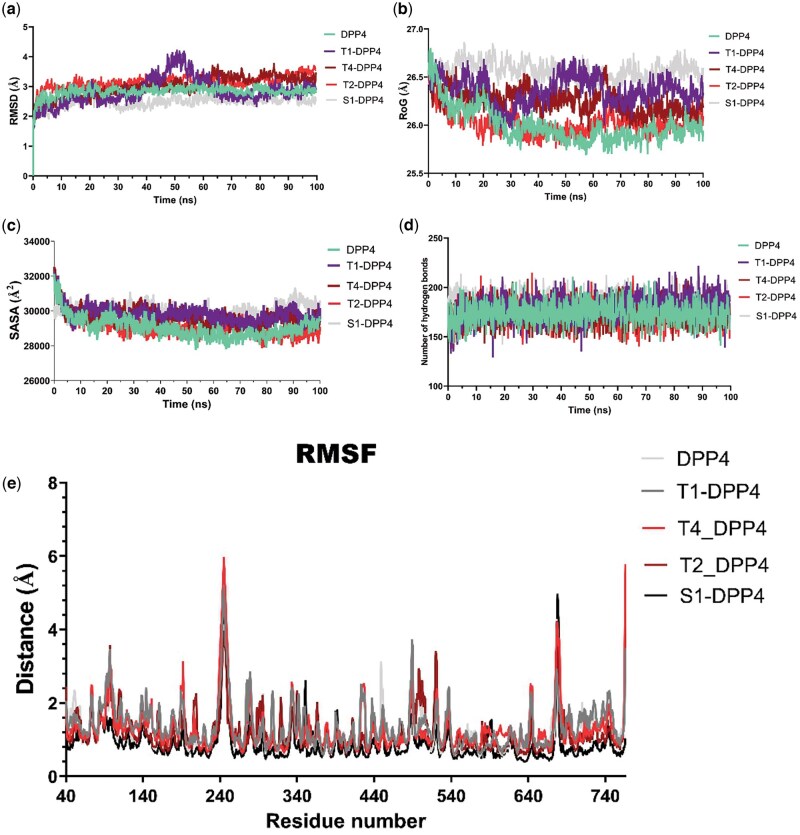
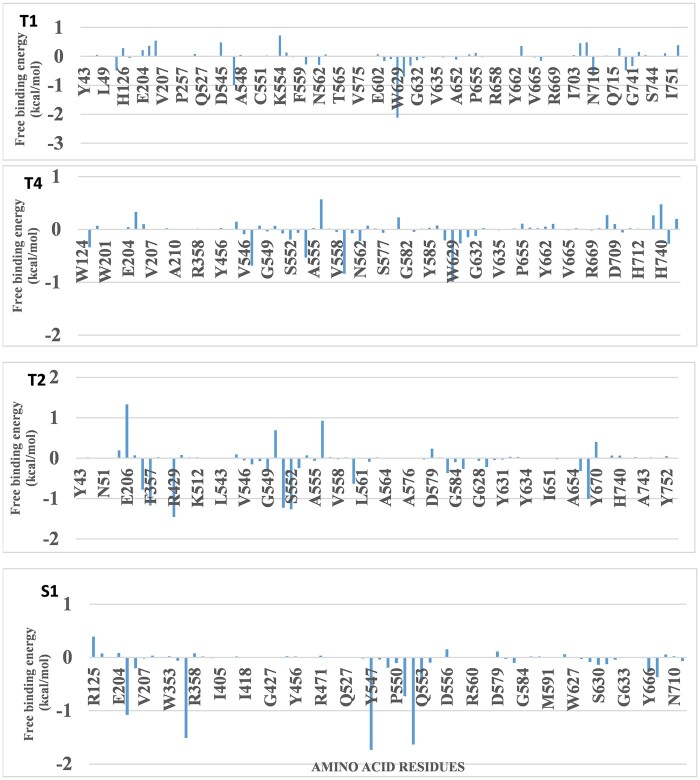
Using molecular docking, molecular dynamics simulations, molecular mechanics with generalized Born and surface area solvation-free energy, and density functional theory analyses, the study revealed dipeptidyl peptidase-4 as a promising target. Cucurbitacin B, 6-oxoisoiguesterin, and 20-epi-isoiguesterinol were identified as potential dipeptidyl peptidase-4 inhibitors with strong binding affinities. These triterpenoids interacted with key catalytic and hydrophobic pockets of dipeptidyl peptidase-4, demonstrating structural stability and flexibility under dynamic conditions, as indicated by dynamics simulation parameters. The free energy analysis further supported the binding affinities in dynamic environments. Quantum mechanical calculations revealed favorable highest occupied molecular orbital and lowest unoccupied molecular orbital energy profiles, indicating the suitability of the hits as proton donors and acceptors, which likely enhance their molecular interactions with the targets. Moreover, the terpenoids showed desirable drug-like properties, suggesting their potential as safe and effective dipeptidyl peptidase-4 inhibitors. These findings may pave the way for the development of novel antidiabetic agents and nutraceuticals based on these promising hits.
Not applicable.
研究新型药物 - 靶点相互作用对于拓展人类疾病中新兴治疗靶点的化学空间至关重要。在此,我们探索了二肽基肽酶 - 4和蛋白酪氨酸磷酸酶1B与非洲抗糖尿病植物中选定萜类化合物的相互作用。
通过分子对接、分子动力学模拟、广义玻恩表面面积溶剂化自由能分子力学以及密度泛函理论分析,该研究表明二肽基肽酶 - 4是一个有前景的靶点。葫芦素B、6 - 氧代异绞股蓝酯苷和20 - 表 - 异绞股蓝醇被鉴定为具有强结合亲和力的潜在二肽基肽酶 - 4抑制剂。这些三萜类化合物与二肽基肽酶 - 4的关键催化和疏水口袋相互作用,动力学模拟参数表明它们在动态条件下具有结构稳定性和灵活性。自由能分析进一步支持了在动态环境中的结合亲和力。量子力学计算揭示了有利的最高占据分子轨道和最低未占据分子轨道能量分布,表明这些命中化合物作为质子供体和受体的适宜性,这可能增强它们与靶点的分子相互作用。此外,这些萜类化合物表现出理想的类药物性质,表明它们作为安全有效的二肽基肽酶 - 4抑制剂的潜力。这些发现可能为基于这些有前景的命中化合物开发新型抗糖尿病药物和营养保健品铺平道路。
不适用。