St Clair-Glover Mitchell, Yousuf Arsalan, Kaul Dominic, Dottori Mirella, Adams David J
Molecular Horizons, Faculty of Science, Medicine and Health, University of Wollongong, Wollongong, New South Wales, Australia.
Sydney Pharmacy School, Faculty of Medicine and Health, The University of Sydney, Sydney, New South Wales, Australia.
J Neurochem. 2025 Jan;169(1):e70004. doi: 10.1111/jnc.70004.
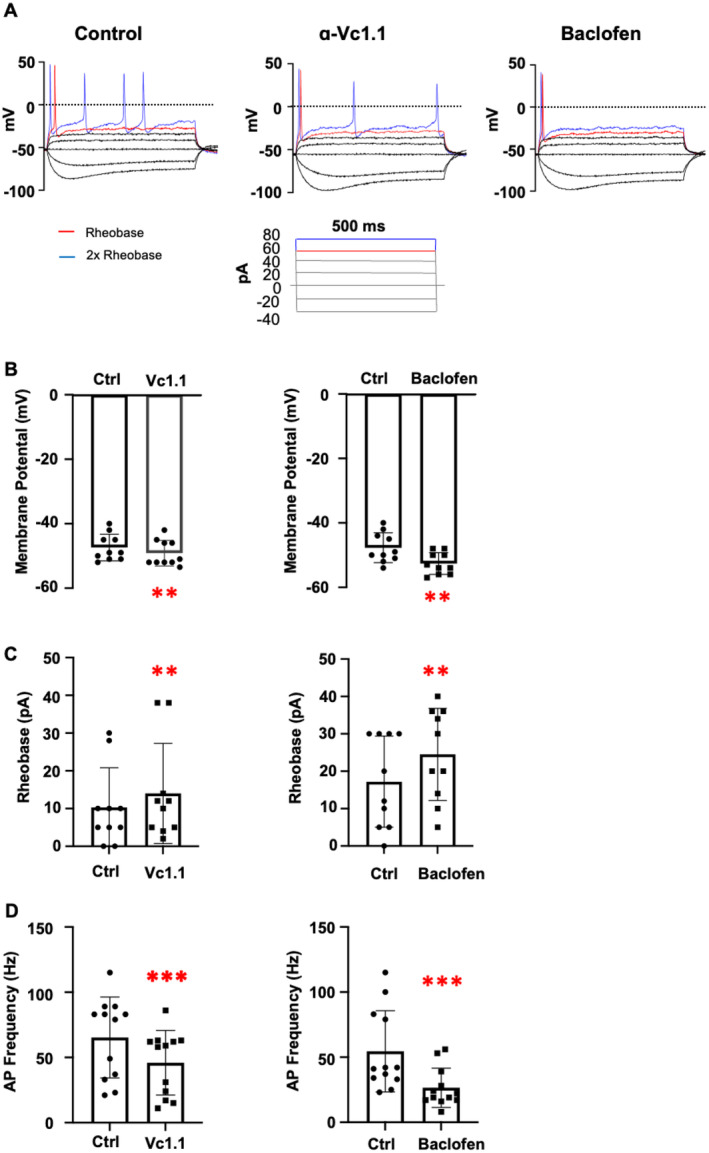
GABA receptor (GABAR) activation is known to alleviate pain by reducing neuronal excitability, primarily through inhibition of high voltage-activated (HVA) calcium (Ca2.2) channels and potentiating G protein-coupled inwardly rectifying potassium (GIRK) channels. Although the analgesic properties of small molecules and peptides have been primarily tested on isolated murine dorsal root ganglion (DRG) neurons, emerging strategies to develop, study, and characterise human pluripotent stem cell (hPSC)-derived sensory neurons present a promising alternative. In this study, hPSCs were efficiently differentiated into peripheral DRG-induced sensory neurons (iSNs) using a combined chemical and transcription factor-driven approach via a neural crest cell intermediate. Molecular characterisation and transcriptomic analysis confirmed the expression of key DRG markers such as BRN3A, ISLET1, and PRPH, in addition to GABAR and ion channels including Ca2.2 and GIRK1 in iSNs. Functional characterisation of GABAR was conducted using whole-cell patch clamp electrophysiology, assessing neuronal excitability under current-clamp conditions in the absence and presence of GABAR agonists baclofen and α-conotoxin Vc1.1. Both baclofen (100 μM) and Vc1.1 (1 μM) significantly reduced membrane excitability by hyperpolarising the resting membrane potential and increasing the rheobase for action potential firing. In voltage-clamp mode, baclofen and Vc1.1 inhibited HVA Ca channel currents, which were attenuated by the selective GABAR antagonist CGP 55845. However, modulation of GIRK channels by GABARs was not observed in the presence of baclofen or Vc1.1, suggesting that functional GIRK1/2 channels were not coupled to GABARs in hPSC-derived iSNs. This study is the first to report GABAR modulation of membrane excitability in iSNs by baclofen and Vc1.1, highlighting their potential as a future model for studying analgesic compounds.
已知γ-氨基丁酸受体(GABAR)激活主要通过抑制高电压激活(HVA)钙(Ca2.2)通道和增强G蛋白偶联内向整流钾(GIRK)通道来降低神经元兴奋性,从而减轻疼痛。尽管小分子和肽的镇痛特性主要在分离的小鼠背根神经节(DRG)神经元上进行了测试,但开发、研究和表征人多能干细胞(hPSC)衍生的感觉神经元的新策略提供了一个有前景的替代方案。在本研究中,通过神经嵴细胞中间体,采用化学和转录因子驱动相结合的方法,将hPSC高效分化为外周DRG诱导的感觉神经元(iSNs)。分子表征和转录组分析证实了iSNs中关键DRG标志物如BRN3A、ISLET1和PRPH的表达,以及GABAR和包括Ca2.2和GIRK1在内的离子通道的表达。使用全细胞膜片钳电生理学对GABAR进行功能表征,在电流钳条件下,评估在不存在和存在GABAR激动剂巴氯芬和α-芋螺毒素Vc1.1的情况下神经元的兴奋性。巴氯芬(100μM)和Vc1.1(1μM)均通过使静息膜电位超极化和增加动作电位发放的阈强度,显著降低了膜兴奋性。在电压钳模式下,巴氯芬和Vc1.1抑制了HVA钙通道电流,该电流被选择性GABAR拮抗剂CGP 55845减弱。然而,在存在巴氯芬或Vc1.1的情况下,未观察到GABAR对GIRK通道的调节作用,这表明在hPSC衍生的iSNs中,功能性GIRK1/2通道未与GABAR偶联。本研究首次报道了巴氯芬和Vc1.1对iSNs膜兴奋性的GABAR调节作用,突出了它们作为未来研究镇痛化合物模型的潜力。