Zhang Lei, Hall Gentzon, Han Peitong, Li Chunzhen, Cui Jieyuan
Department of Pediatric Nephrology, Children's Hospital of Hebei Province Affiliated to Hebei Medical University, Shijiazhuang, China.
Duke University School of Medicine, Duke Molecular Physiology Institute, Durham, NC, United States.
Front Pediatr. 2025 Jan 14;12:1378083. doi: 10.3389/fped.2024.1378083. eCollection 2024.
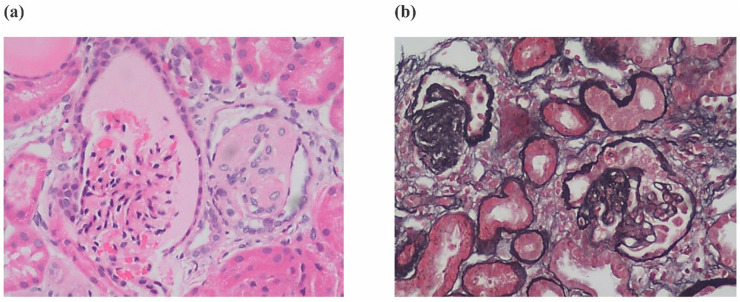
Primary coenzyme Q10 (CoQ10) deficiency is an autosomal recessive genetic disease caused by mitochondrial dysfunction. Variants in Coenzyme Q8B () can cause primary CoQ10 deficiency. -related glomerulopathy is a recently recognized glomerular disease that most often presents as steroid-resistant nephrotic syndrome (SRNS) in childhood. The disease often progresses to kidney failure and the renal histopathology is most commonly focal segmental glomerulosclerosis (FSGS).
Four SRNS cases (2 females and 2 males) from 2 unrelated families who were followed clinically for nearly 3 years. Clinical exome testing and analyses were performed by MyGenostics Laboratory in China to evaluate unexplained proteinuria given the strong family history of glomerular disease and histologic evidence of SRNS. Pathogenic variants were identified in in the exome studies and confirmed by direct sequencing.
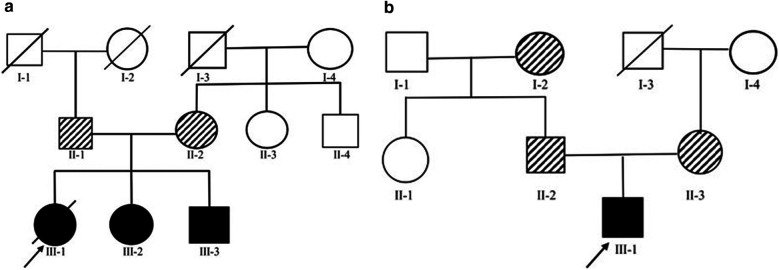
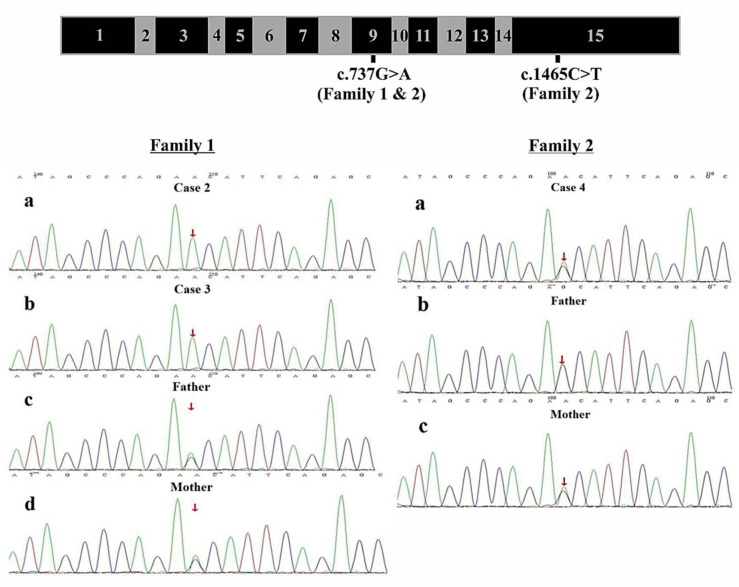
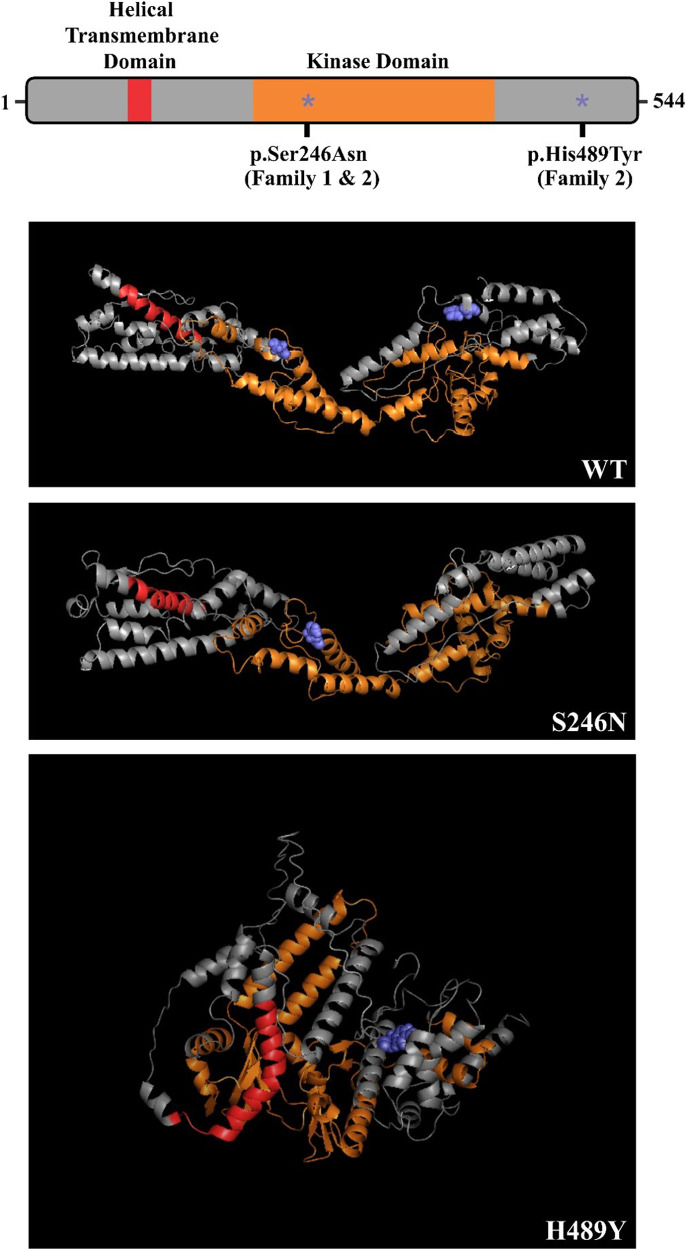
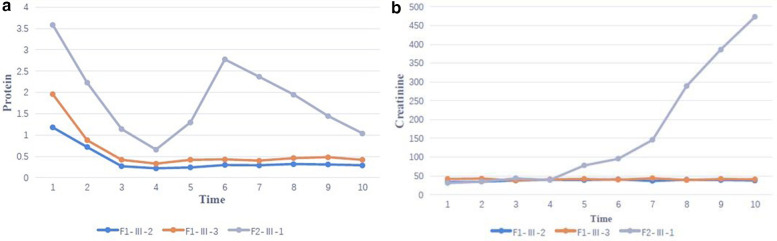
Clinical exome sequencing revealed biallelic variants of the gene in 2 families. In the Family 1, the oldest of three affected siblings died of renal failure at 11 years of age. Based on the results of genetic testing which identified a homozygous variant of , the other two affected siblings with mild proteinuria and normal renal function were treated with CoQ10 oral supplementation at an early stage. Coenzyme Q10 treatment was effective in reducing proteinuria levels in both patients from Family 1 over the first 6 months and the two patients still have low-level proteinuria and normal renal function at nearly three years. In Family 2, clinical exome sequencing revealed a compoundheterozygous variants of in a patient with biopsy- proven FSGS. His disease was unresponsive to prior treatment with glucocorticoids and cyclosporine. Oral CoQ10 was initiated based on his genetic diagnosis and was it was effective in reducing proteinuria over the first 5 months months of therapy. However after 1 year, his disease progressed tokidney failure. Kidney transplantation was performed at 5 years of age and his condition has been stable without rejection and no recurrence of disease.
gene variant-related glomerulopathy often presents as SRNS without obvious extrarenal manifestations. The histopathology is mainly FSGS and follows an autosomal recessive mode of inheritance. Some patients may benefit from early coenzyme Q10 supplementation. For patients whose disease progresses to kidney failure, kidney transplantation can be an effective treatment. For children with unexplained proteinuria and abnormal renal function, genetic testing should be performed early in the course of disease to guide therapy where possible and improve prognosis.
原发性辅酶Q10(CoQ10)缺乏症是一种由线粒体功能障碍引起的常染色体隐性遗传病。辅酶Q8B()基因变异可导致原发性CoQ10缺乏症。与相关的肾小球病是一种最近才被认识的肾小球疾病,在儿童期最常表现为激素抵抗型肾病综合征(SRNS)。该疾病常进展为肾衰竭,肾脏组织病理学最常见的是局灶节段性肾小球硬化(FSGS)。
对来自2个无关家庭的4例SRNS病例(2名女性和2名男性)进行了近3年的临床随访。鉴于肾小球疾病的家族史及SRNS的组织学证据,中国的迈基诺基因检测实验室进行了临床外显子组检测和分析,以评估不明原因的蛋白尿。在外显子组研究中鉴定出致病变异,并通过直接测序进行了确认。
临床外显子组测序在2个家庭中发现了基因的双等位基因变异。在家庭1中,3名受影响的兄弟姐妹中年龄最大的在11岁时死于肾衰竭。基于基因检测结果,该结果鉴定出了的纯合变异,另外两名有轻度蛋白尿且肾功能正常的受影响兄弟姐妹在早期接受了辅酶Q10口服补充治疗。辅酶Q10治疗在最初6个月内有效降低了家庭1中两名患者的蛋白尿水平,近3年时这两名患者仍有低水平蛋白尿且肾功能正常。在家庭2中,临床外显子组测序在一名经活检证实为FSGS的患者中发现了的复合杂合变异。他的疾病对先前的糖皮质激素和环孢素治疗无反应。基于他的基因诊断开始口服辅酶Q10,在治疗的前5个月有效降低了蛋白尿。然而1年后,他的疾病进展为肾衰竭。在5岁时进行了肾移植,他的病情一直稳定,无排斥反应且疾病无复发。
基因变异相关的肾小球病常表现为SRNS,无明显肾外表现。组织病理学主要为FSGS,呈常染色体隐性遗传模式。一些患者可能从早期补充辅酶Q10中获益。对于疾病进展为肾衰竭的患者,肾移植可能是一种有效的治疗方法。对于不明原因蛋白尿且肾功能异常的儿童,应在病程早期进行基因检测,以便尽可能指导治疗并改善预后。