Seely J E, Persson L, Sertich G J, Pegg A E
Biochem J. 1985 Mar 1;226(2):577-86. doi: 10.1042/bj2260577.
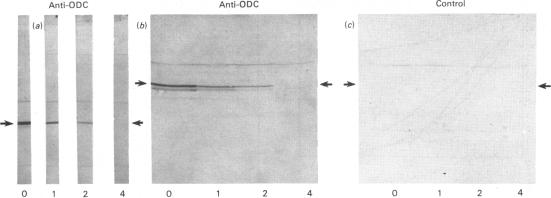
Comparisons were made of ornithine decarboxylase isolated from Morris hepatoma 7777, thioacetamide-treated rat liver and androgen-stimulated mouse kidney. The enzymes from each source were purified in parallel and their size, isoelectric point, interaction with a monoclonal antibody or a monospecific rabbit antiserum to ornithine decarboxylase, and rates of inactivation in vitro, were studied. Mouse kidney, which is a particularly rich source of ornithine decarboxylase after androgen induction, contained two distinct forms of the enzyme which differed slightly in isoelectric point, but not in Mr. Both forms had a rapid rate of turnover, and virtually all immunoreactive ornithine decarboxylase protein was lost within 4h after protein synthesis was inhibited. Only one form of ornithine decarboxylase was found in thioacetamide-treated rat liver and Morris hepatoma 7777. No differences between the rat liver and hepatoma ornithine decarboxylase protein were found, but the rat ornithine decarboxylase could be separated from the mouse kidney ornithine decarboxylase by two-dimensional gel electrophoresis. The rat protein was slightly smaller and had a slightly more acid isoelectric point. Studies of the inactivation of ornithine decarboxylase in vitro in a microsomal system [Zuretti & Gravela (1983) Biochim. Biophys. Acta 742, 269-277] showed that the enzymes from rat liver and hepatoma 7777 and mouse kidney were inactivated at the same rate. This inactivation was not due to degradation of the enzyme protein, but was probably related to the formation of inactive forms owing to the absence of thiol-reducing agents. Treatment with 1,3-diaminopropane, which is known to cause an increase in the rate of degradation of ornithine decarboxylase in vivo [Seely & Pegg (1983) Biochem. J. 216, 701-717] did not stimulate inactivation by microsomal extracts, indicating that this system does not correspond to the rate-limiting step of enzyme breakdown in vivo.
对从莫里斯肝癌7777、硫代乙酰胺处理的大鼠肝脏以及雄激素刺激的小鼠肾脏中分离出的鸟氨酸脱羧酶进行了比较。对每种来源的酶进行了平行纯化,并研究了它们的大小、等电点、与鸟氨酸脱羧酶单克隆抗体或单特异性兔抗血清的相互作用以及体外失活速率。小鼠肾脏在雄激素诱导后是鸟氨酸脱羧酶特别丰富的来源,其中含有两种不同形式的酶,它们的等电点略有不同,但分子量相同。两种形式的酶都有快速的周转速率,并且在蛋白质合成被抑制后的4小时内,几乎所有免疫反应性鸟氨酸脱羧酶蛋白都消失了。在硫代乙酰胺处理的大鼠肝脏和莫里斯肝癌7777中仅发现一种形式的鸟氨酸脱羧酶。大鼠肝脏和肝癌的鸟氨酸脱羧酶蛋白之间没有差异,但大鼠鸟氨酸脱羧酶可以通过二维凝胶电泳与小鼠肾脏鸟氨酸脱羧酶分离。大鼠蛋白略小,等电点略偏酸性。在微粒体系统中对鸟氨酸脱羧酶体外失活的研究[祖雷蒂和格拉韦拉(1983年)《生物化学与生物物理学报》742,269 - 277]表明,大鼠肝脏、肝癌7777和小鼠肾脏中的酶以相同的速率失活。这种失活不是由于酶蛋白的降解,而是可能与由于缺乏硫醇还原剂而形成无活性形式有关。已知1,3 - 二氨基丙烷会导致体内鸟氨酸脱羧酶降解速率增加[西利和佩格(1983年)《生物化学杂志》216,701 - 717],但它不会刺激微粒体提取物的失活,这表明该系统与体内酶分解的限速步骤不对应。