Fisher Rachael P, Matheny Lindsay, Ankeny Sarrah, Qin Liya, Coleman Leon G, Vetreno Ryan P
Bowles Center for Alcohol Studies, School of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States.
Department of Pharmacology, School of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States.
Front Aging Neurosci. 2025 Feb 19;17:1531628. doi: 10.3389/fnagi.2025.1531628. eCollection 2025.
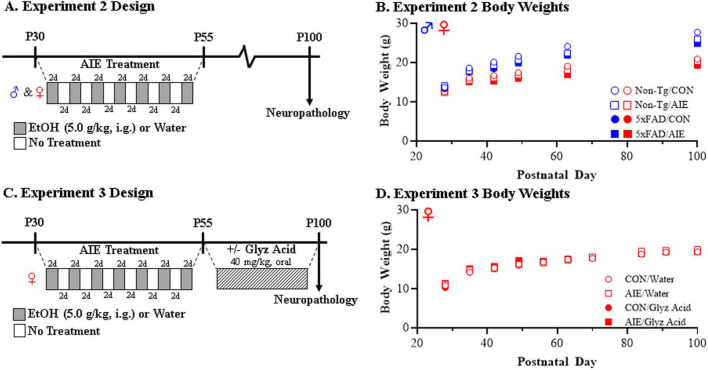
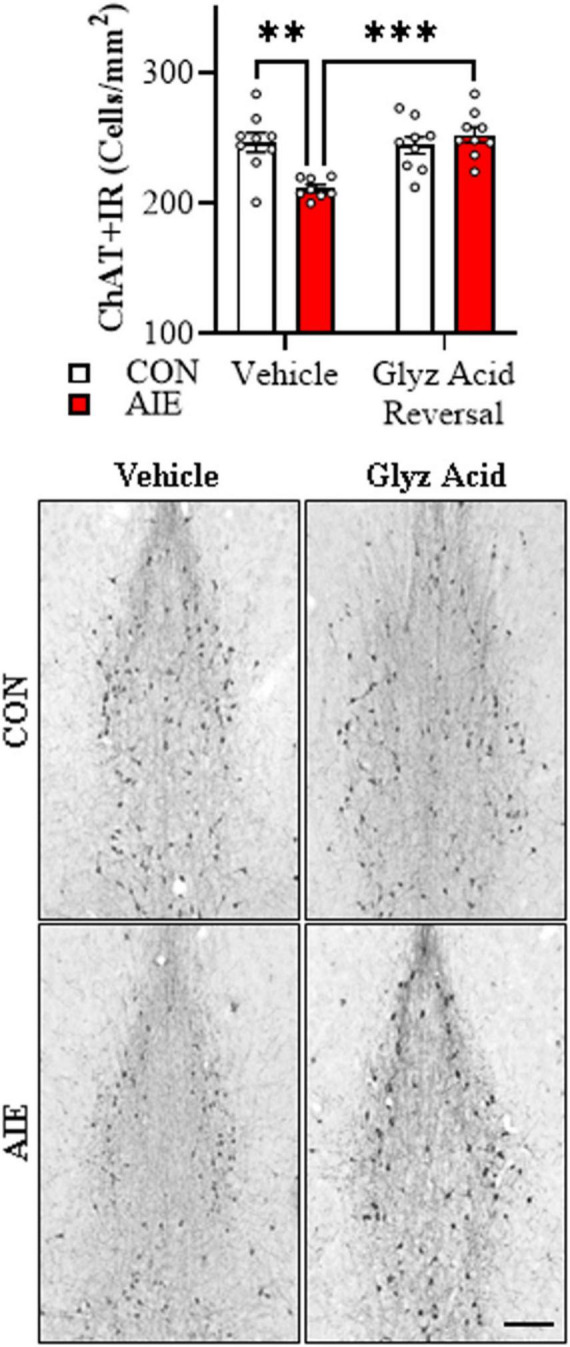
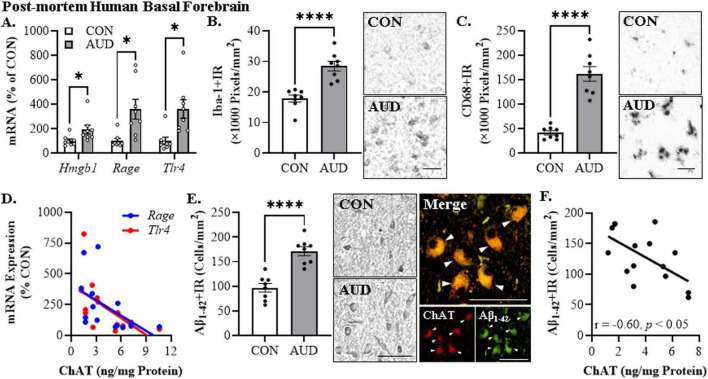
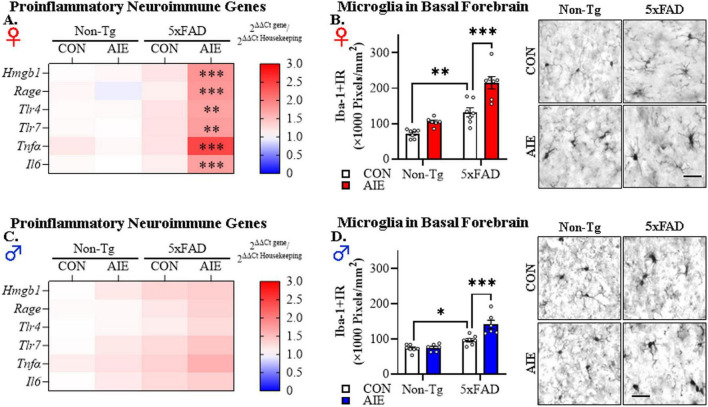
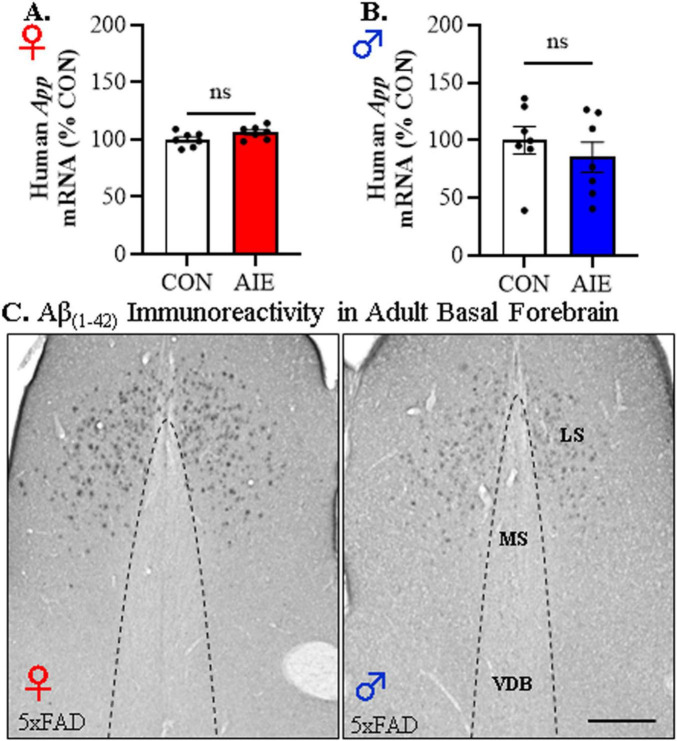
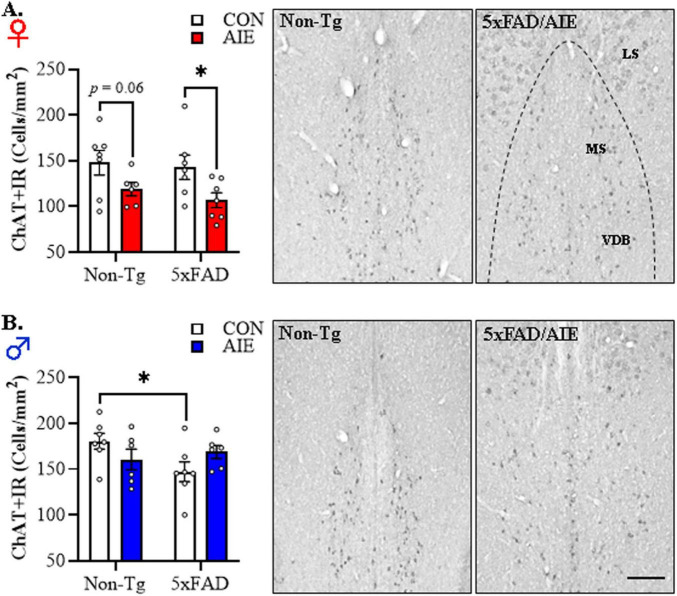
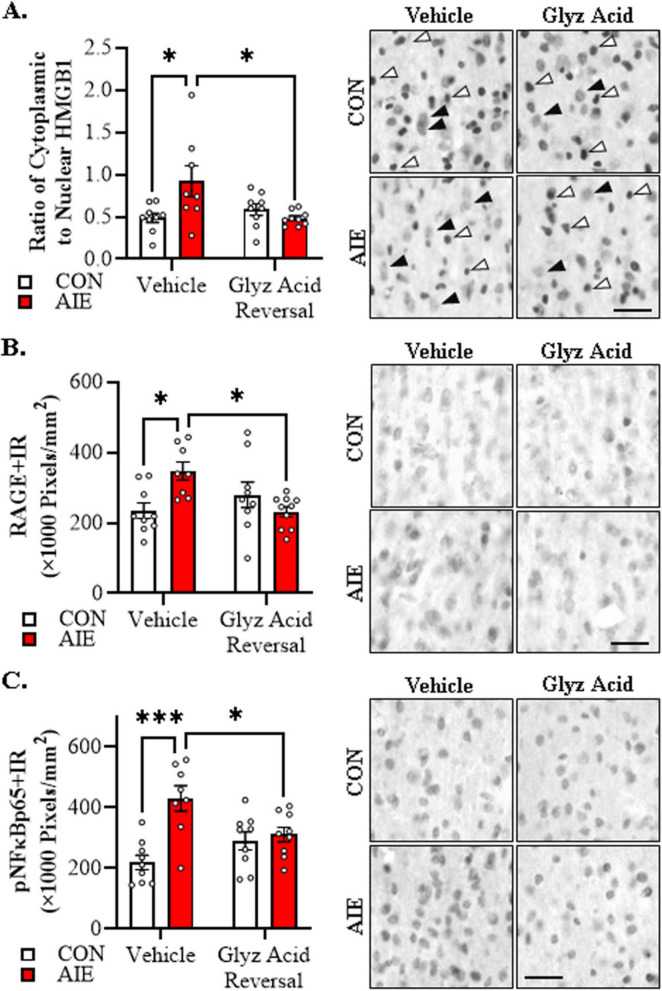
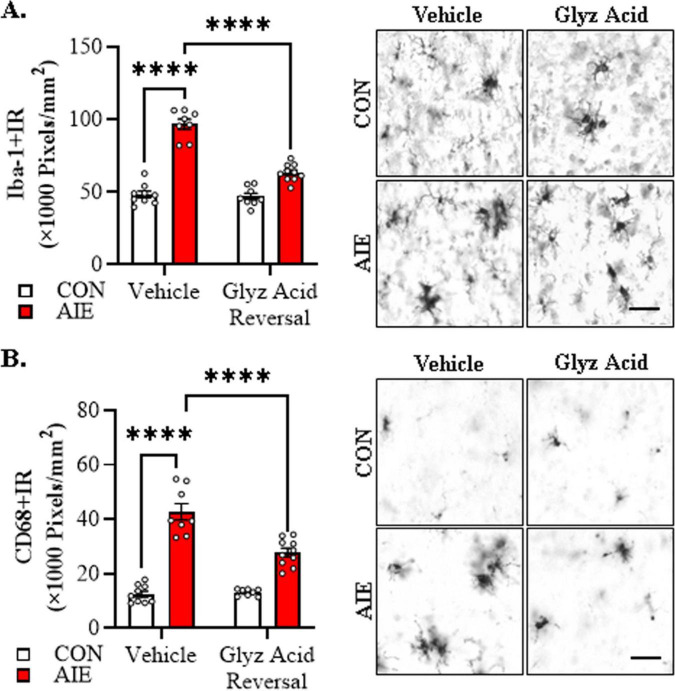
Human studies suggest that heavy alcohol use may be an etiological factor contributing to the development of Alzheimer's disease (AD) neuropathology. Both alcohol use disorder (AUD) and AD share common underlying neuropathology, including proinflammatory high-mobility group box 1 (HMGB1)-mediated neuroimmune signaling and basal forebrain cholinergic neuron degeneration. Adolescent onset of binge drinking represents a significant risk factor for later development of an AUD, and accumulating evidence suggests that adolescent initiation of heavy alcohol use induces HMGB1 signaling and causes degeneration of the basal forebrain cholinergic system that persists into adulthood. However, it is unknown whether adolescent binge drinking confers increased risk for later development of AD-associated neuropathology through persistent induction of proinflammatory HMGB1 neuroimmune signaling. To investigate this question, we first (Experiment 1) assessed AD-associated neuropathology in the post-mortem human basal forebrain of individuals with AUD and an adolescent age of drinking onset relative to age-matched moderate drinking controls (CONs). In Experiment 2, we treated non-transgenic and 5xFAD male and female mice, which overexpress both mutant human APP and PS1, with adolescent intermittent ethanol (AIE; 5.0 g/kg, i.g. 2-days on/2-days off; postnatal day [P]30 - P55), and assessed AD-associated neuropathology in the adult (P100) basal forebrain. In Experiment 3, 5xFAD female mice received AIE treatment followed by glycyrrhizic acid (150 mg/L), an HMGB1 inhibitor, in drinking water from P56 to P100, and basal forebrain tissue was collected on P100 for assessment of AD-associated neuropathology. In the post-mortem human AUD basal forebrain (Experiment 1), we report upregulation of and the HMGB1 receptors and as well as microglial activation and increased intraneuronal Aβ accumulation in association with reduced cholinergic neuron marker expression (ChAT). In the 5xFAD mouse model (Experiment 2), AIE accelerated AD-associated induction of proinflammatory neuroimmune genes, microglial activation, and reductions of ChAT+ basal forebrain cholinergic neurons in the adult female, but not male, basal forebrain. In Experiment 3, post-AIE treatment with glycyrrhizic acid rescued the AIE-induced acceleration of AD-associated increases in proinflammatory HMGB1 neuroimmune signaling, microglial activation, and persistent reductions of basal forebrain cholinergic neurons in adult 5xFAD female mice. Together, these findings suggest that adolescent binge ethanol exposure may represent an underappreciated etiological factor contributing to onset of AD-associated neuropathology in adulthood through HMGB1- mediated neuroimmune signaling.
人体研究表明,大量饮酒可能是导致阿尔茨海默病(AD)神经病理学发展的一个病因。酒精使用障碍(AUD)和AD具有共同的潜在神经病理学特征,包括促炎的高迁移率族蛋白B1(HMGB1)介导的神经免疫信号传导和基底前脑胆碱能神经元变性。青少年时期开始的暴饮代表了日后发生AUD的一个重要危险因素,越来越多的证据表明,青少年开始大量饮酒会诱导HMGB1信号传导,并导致基底前脑胆碱能系统变性,这种变性会持续到成年期。然而,尚不清楚青少年暴饮是否会通过持续诱导促炎的HMGB1神经免疫信号传导而增加日后发生AD相关神经病理学的风险。为了研究这个问题,我们首先(实验1)评估了AUD患者以及青少年饮酒起始年龄组的尸检人类基底前脑中与AD相关的神经病理学,与年龄匹配的适度饮酒对照组(CONs)进行比较。在实验2中,我们用青少年间歇性乙醇(AIE;5.0 g/kg,灌胃,2天饮用/2天不饮用;出生后第[P]30 - P55天)处理同时过表达突变型人类APP和PS1的非转基因和5xFAD雄性和雌性小鼠,并评估成年(P100)基底前脑中与AD相关的神经病理学。在实验3中,5xFAD雌性小鼠接受AIE处理,然后从P56到P100在饮用水中给予甘草酸(150 mg/L),一种HMGB1抑制剂,并在P100收集基底前脑组织以评估与AD相关的神经病理学。在尸检的人类AUD基底前脑(实验1)中,我们报告了与胆碱能神经元标记物表达(ChAT)降低相关的 以及HMGB1受体 和 的上调,以及小胶质细胞活化和神经元内Aβ积累增加。在5xFAD小鼠模型(实验2)中,AIE加速了成年雌性而非雄性基底前脑中与AD相关的促炎神经免疫基因的诱导、小胶质细胞活化以及ChAT+基底前脑胆碱能神经元的减少。在实验3中,AIE处理后用甘草酸挽救了AIE诱导的成年5xFAD雌性小鼠中与AD相关的促炎HMGB1神经免疫信号传导增加、小胶质细胞活化以及基底前脑胆碱能神经元持续减少的加速。总之,这些发现表明,青少年暴饮乙醇暴露可能是一个未被充分认识的病因,通过HMGB1介导的神经免疫信号传导促成成年期AD相关神经病理学的发生。