Panahi Bahman, Hamid Rasmieh
Department of Genomics, Branch for Northwest & West Region, Agricultural Biotechnology Research Institute of Iran (ABRII), Agricultural Research, Education and Extension Organization (AREEO), Tabriz, 5156915-598, Iran.
Department of Plant Breeding, Cotton Research Institute of Iran (CRII), Agricultural Research, Education and Extension Organization (AREEO), Gorgan, Iran.
Biochem Biophys Rep. 2025 Feb 20;41:101958. doi: 10.1016/j.bbrep.2025.101958. eCollection 2025 Mar.
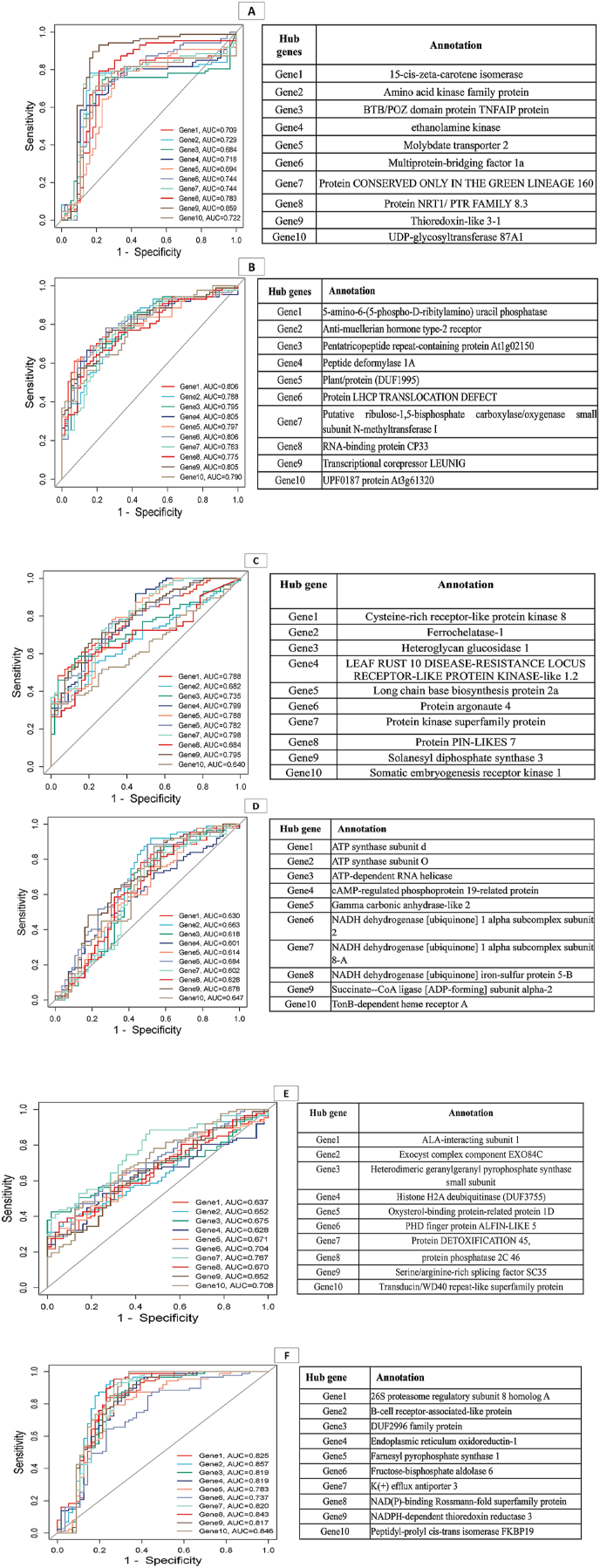
Fungal infections pose a considerable threat to the cultivation of barley () and often limit the crop yield. During infection, the transcriptome undergoes extensive reprogramming involving several regulatory pathways. To address this complexity, we performed a comprehensive meta-analysis and co-expression network analysis using rigorously curated RNA-seq datasets from three different fungal diseases. Pre-processing of the data, including batch effect correction, ensured high-quality integration of the datasets. Module-trait relationship (MTR) analysis identified functional modules associated with fungal disease response. Hub genes within these modules were prioritized by multi-model centrality analyses using Cytoscape, which considered the metrics Degree, Closeness, Betweenness and Maximum Clique Centrality together with the MCODE algorithm to detect densely connected subclusters. These hub genes were further validated by cross-validation and receiver operating characteristic (ROC) curve analysis and achieved AUC values greater than 0.7, confirming their robustness. A total of 6688 consistently expressed genes were identified, including 879 upregulated and 701 downregulated genes. Co-expression networks revealed 19 different gene modules, six of which were significantly associated with the response of barley to fungal infection. The blue module in particular was associated with immune responses such as activation of the MAPK cascade and pathogen recognition, while the green module correlated with defence mechanisms and secondary metabolism. The hub genes within these modules showed high predictive power for fungal resistance, as shown by the AUC values of the ROC curve of over 0.7, emphasizing their potential as biomarkers. This study uniquely integrates multiple RNA-seq datasets to identify novel regulatory networks and hub genes, including 345 transcription factors (TFs) from different families, with MYB and bHLH being particularly abundant. The results provide valuable insights into regulatory networks associated with fungal disease response in barley. These results can support genomic selection and marker-assisted breeding programs and accelerate the development of resistant varieties.
真菌感染对大麦种植构成了相当大的威胁,并且常常限制作物产量。在感染过程中,转录组会经历广泛的重编程,涉及多个调控途径。为了解决这种复杂性,我们使用来自三种不同真菌病害的经过严格筛选的RNA测序数据集进行了全面的荟萃分析和共表达网络分析。数据的预处理,包括批次效应校正,确保了数据集的高质量整合。模块-性状关系(MTR)分析确定了与真菌病害反应相关的功能模块。使用Cytoscape通过多模型中心性分析对这些模块内的枢纽基因进行了优先级排序,该分析考虑了度、接近度、介数和最大团中心性等指标以及MCODE算法来检测紧密连接的子簇。这些枢纽基因通过交叉验证和受试者工作特征(ROC)曲线分析进一步验证,AUC值大于0.7,证实了它们的稳健性。总共鉴定出6688个持续表达的基因,其中包括879个上调基因和701个下调基因。共表达网络揭示了19个不同的基因模块,其中六个与大麦对真菌感染的反应显著相关。特别是蓝色模块与免疫反应相关,如MAPK级联的激活和病原体识别,而绿色模块与防御机制和次生代谢相关。这些模块内的枢纽基因对真菌抗性显示出高预测能力,ROC曲线的AUC值超过0.7就表明了这一点,强调了它们作为生物标志物的潜力。本研究独特地整合了多个RNA测序数据集,以识别新的调控网络和枢纽基因,包括来自不同家族的345个转录因子(TFs),其中MYB和bHLH特别丰富。这些结果为与大麦真菌病害反应相关的调控网络提供了有价值的见解。这些结果可以支持基因组选择和标记辅助育种计划,并加速抗性品种的开发。